











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink1.Introduction
Aluminum nitride (AlN), an III-V compound semiconductor displays the wurtzite phase as the ground state, however, the cubic zinc blende and rock salt (sodium chloride) are present as metastable geometries. In the wurtzite and zinc blende configurations, the compound is tetrahedrally coordinated, where each atom forms four equal bonds. The hexagonal wurtzite structure has an experimentally determined direct bandgap of 6.2 eV, which is the largest bandgap in the III-V semiconductors 1. Besides it has high thermal conductivity (2.85 W/cmK) and a small thermal coefficient 2,3, so this material is suitable for devices used at high temperatures 4. The wurtzite phase experimental lattice parameters are: a = b= 3.11 Å and c = 4.98 Å5-6, and the calculated are a = b = 3.13 Å and c = 4.97 Å. The metastable zincblende phase has an indirect band-gap of 4.9 eV, even larger than some III-V semiconductors (e.g., GaN, 3.4 eV, and InN, 1.9 eV) 7.
Schulz et al 8 have used X-ray spectroscopy to measure the AlN and GaN wurtzite phase lattice parameters. Results yielded a = 3.110 A, c = 4.980 A, u = 0.3821 for AlN and a = 3.190 A, c = 5.189 A, u = 0.377 for GaN. Mention must be done that according to results, AlN measured parameters deviate from the ideal parameters; in contrast to those corresponding to GaN, which are close to the ideal values. Yadav et al.,9 have demonstrated experimentally the existence of the zincblende metastable phase for AlN thin films. Moreover, under uniaxial indentation, zincblende-AlN partially transforms to wurtzite-AlN. Experiments yield for AlN in the wurtzite phase a = 3.11 A and c = 4.98 A. Therefore, the wurtzite structure remains to be the ground state. DFT calculations have shown that the wurtzite phase is the ground state structure, in agreement with the experiment, with the zincblende and rock salt as metastable phases.
Recent studies have predicted that AlN doped with nickel (Ni) may exhibit magnetic
properties, even it can display ferromagnetic behavior above room temperature. Other
studies have been reported on the chromium doped AlN films with a Curie temperature
(Tc) higher than 900 K and copper doped AlN films with
The emphasis of this work is to investigate the electronic properties and phase transitions of AlN. Density functional theory (DFT) has been applied to explore the possible structural phase transitions as induced by applied pressure. It is well known that the DFT theory underestimates the semiconductor energy gap value in either GGA or LDA approaches. In the present work, we include the modified Becke-Johnson theory to improve the energy gap value calculation. In fact, results show better agreement with experimental data. The report is organized as follows: Section 2 describes the methodology; Section 3 is devoted to presenting results and discussion. Finally, 4 is for conclusions.
2.Computational method
The studies were done within the periodic density functional theory as implemented in the WIEN2k code 19. The electron-ion interactions are treated according to the full-potential model. The electron states are expanded in a linear combination of plane waves and atomic orbitals. Two approaches are invoked: the generalized gradient approximation (GGA) and the local density approximation (LDA). Calculations were performed using the Full-Potential Linearized Augmented Plane-Wave (FP-LAPW) method to solve the Kohn-Sham equations, within the periodic density functional theory (DFT) 8, as implemented in the Wien2k code 19. The exchange-correlation potential energy was treated according to the generalized gradient approximation (GGA) with the Perdew, Burke, and Enzernof (PBE) functional 20 and the local density approximation (LDA). Additionally, the GGA and LDA approaches were used in combination with the Becke-Johnson exchange potential 21-23 in order to determine, in a more accurate fashion, the AlN electronic bandgap. In this work, the core electron states and valence states were treated separately, using a cut-off energy of -6.0 Ryd. Different k-points were used, which depends on the structural phase to generate a k-mesh to integrate Kohn-Sham equations in the first Brillouin zone. The convergence criteria for self-consistent iterations are: 0.0001 Ryd for the energy and 0.001e for the charge. In the interstitial region, the electron wave function was expanded in plane waves and inside the muffin-tin as a linear combination of atomic orbitals. The form used for the electron wave function was as:
where
In the structural construction, 2 Al and 2 N atoms compose the AlN wurtzite unit cell, 1 Al and 1 N atom form the zinc blende and NaCl unit cells 4. The calculated lattice parameters of the wurtzite structure are: a = b = 3.0844 Å and c = 4.96 Å, which compare well with the experimental data 18,19 (a = b = 3.11 Å and c = 4.98 Å).
3.Results
The total energy of the relaxed structures and energies of the isolated atoms that form the wurtzite, zinc blende, and rock salt (NaCl) crystal structures (see Fig.1 for all three phases), were used to determine the cohesion energy according to the formula
where Ec is the cohesion energy, ES is the energy of every structural phase, EAl and EN are the energies of the isolated atoms.
The Murnaghan formalism was used to fit the calculated curves as given by the equation
where B0 and B´0 are the bulk modulus and its derivative, respectively, V0 is the volume, E0 is the total energy. Both GGA and LDA were compared, in the calculations to determine the total energy in the three phases, as plotted in Fig. 2. Optimized lattice parameters are summarized in Table I. B0 and B´0 are also listed in Table 1.
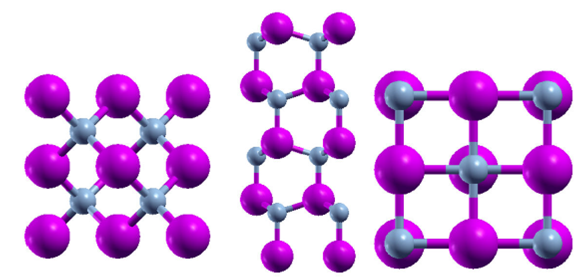
Figure 1 The ALN crystal structures: left zinc blende (ZB), middle wurtzite (WZ) and right rock salt (rs) (NaCl).
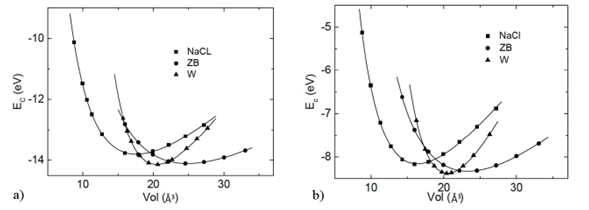
Figure 2 The figure shows the cohesion energy as a function of the primitive unit cell volume. On the left panel, results correspond to the GGA theory and on the right to the LDA approach. ZB denotes zinc blende, W denotes wurtzite, and NaCl sodium chloride (rock salt).
Table I The table reports a comparison of calculated and available experimental values of lattice parameters and band-gap energy of AlN in wurtzite (wz), zincblende (zb), and rock-salt (rs) phases. B and B´ values are also included.
| GGA | LDA | ||||||
| PHASE | Lattice | Lattice | |||||
| parameters | Band-GAP | parameters | Band-GAP | ||||
| Results (Å) | Reported data (Å) | Results (eV) | Reported data (eV) | Results (Å) | Results (eV) | ||
| zb | a = b = c 4.40 | a = b = c 4.4 [9] | 5.048 (indirect) | 4.9 [7] | a = b = c 4.30 | 5.149 (indirect) | |
| a=b=c | a=b=c | 5.966 | a=b=c | 6.687 | |||
| rs | 4.10 | 4.06 [9] | (indirect) | 4.00 | (indirect) | ||
| a=b | a=b | a=b | |||||
| 3.15 | 3.11 [8] | 3.08 | |||||
| c | c | c | 5.876 | ||||
| wz | 4.98 | 4.98 [8] | 5.391 (direct) | 6.015 [1] | 4.96 | (direct) | |
| B(GPa) | B’ | B(GPa) | B’ | ||||
| zb | 18.12 | 3.011 | 19.82 | 3.026 | |||
| rs | 25.87 | 2.899 | 28.20 | 3.086 | |||
| wz | 78.92 | 3.847 | 84.30 | 3.797 | |||
Cohesion energies Ec as functions of volume are reported in Fig. 2, with the volume being per formula unit. In the left panel, the GGA results are exhibited. Note that the most stable structure corresponds to the hexagonal wurtzite phase. It is also noted that energetically the zinc blende structure is very close to the ground state structure, which indicates that this is a meta-stable configuration. Moreover, the NaCl phase may be also formed as a metastable structure. The graph indicates the possible crossing of the wurtzite and zinc blende curves, and the wurtzite and rock salt curves, which suggest the existence of structural phase transition from wurtzite to zinc blende, or from wurtzite to rock salt phases.
The right panel of Fig. 2 exhibits results obtained within the LDA approximation. Note that the graph depicts similar features as those of the GGA calculations. The point to make is concerned with the possible structural phase transition from hexagonal wurtzite to cubic zinc blende or rock salt (NaCl) phases. This is suggested by curves crossing, with the pressure at the transition being calculated using the enthalpy of formation. The pressure at the transition is estimated using the H vs. P graphs, as shown below.
Mention must be done that LDA calculations are generally not accurate enough to describe the energetics of systems. Generalized gradient approximations (GGA’s) go beyond the LDA and LSD descriptions by including density gradients and improve calculated LDA results significantly. That is, it corrects the energetics calculations. It is expected to compare well with the experimental data.
3.1.Formation enthalpy H
The formation of enthalpy H at arbitrary temperature may be written in the form
where H is the enthalpy of formation, E0 is total energy, T is the temperature, S is the entropy, P is the pressure, and V is the volume. For the system at hand T = 0, therefore the equation reduces to
Plots of this equation considering the wurtzite, zinc blende, and rock salt (NaCl) energy minima and their corresponding volume yield straight lines that cross each other, as shown in Fig. 3 and 4. Enthalpy (eV) is plotted as a function of the pressure (GaP). The point at the curves crossing yields the pressure and enthalpy of the phase transition. Figure 3 depicts results of both GGA and LDA theories of the zinc blende and wurtzite phases.
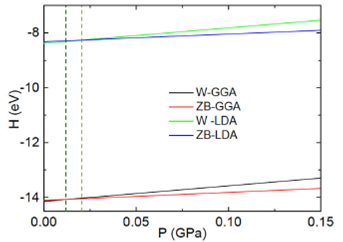
Figure 3 The figure shows the AlN enthalpy (in eV) as a function of the pressure (GaP) of the wurtzite and zinc blende phases. Lower lines are for the GGA approach, and the upper lines correspond to the LDA theory. Vertical dashed lines have been included to point out the line crossing.
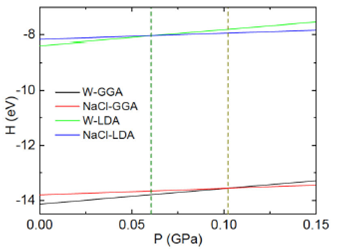
Figure 4 This is to show the AlN enthalpy (in eV) as a function of the pressure (GaP) of the wurtzite and rock salt (NaCl) phases. Lower lines are for the GGA approach, and upper lines correspond to the LDA theory. Vertical dashed lines have been included to point out the line crossing.
Table II Enthalpy and pressure calculation from both theories.
| ENTHALPY | |
| GGA W and ZB | LDA W and ZB |
| H = -14.0727 eV P = 1.215 x 10-2 GPa | H = -8.2620 eV P = 2.086 x 10-2 GPa |
| W and RS | W and RS |
| H = -13.5672 eV P = 1.014 x 10-1 GPa | H = -8.0345 eV P = 6.153 x 10-2 GPa |
Both GGA and LDA approaches yield transition pressures, which are small. This is not a surprise, provided that the minima of the zinc blende structure are quite near those of the wurtzite phase.
Figure 4 shows plots of the enthalpy as a function of pressure in the wurtzite and rock salt (NaCl) geometries. The upper lines are for the LDA calculations and the lower lines correspond to the GGA approach. Similar to 3, energy minima and corresponding volume yield straight lines that cross each other in both approximations. The point at the curves crossing yields the pressure and enthalpy of the phase transition. Both GGA and LDA approaches yield transition pressures, which are small, however larger than those of the wurtzite and zinc blende structures. This is because the minima of the NaCl structure are higher than those of the wurtzite phase.
In this way, for the wurtzite to zinc blende transition the estimated pressures result as: in the GGA 1.215 X 10-2 GPa and the LDA 2.086 X 10-2 GPa. And for the wurtzite to rock salt transition the estimated pressures result as: in the GGA 1.014 X 10-1 GPa and the LDA 6.153 X 10-2 GPa.
3.2.Electronic properties
3.2.1.Band structure
Electronic properties are investigated in terms of the band structure and density of states (DOS). All three-crystal structures are considered: wurtzite, zinc blende, and rock salt, within both GGA and LDA approaches. Mention must be done that these post-processing calculations take into account the modified Becke-Johnson pseudopotentials, which shall yield results in better agreement with experimental data, particularly in the energy gap evaluation.
The first results reported here correspond to the GGA calculations. Figure 5 depicts the band structure in the ground state, which corresponds to the wurtzite phase. Note that this phase exhibits a direct gap semiconductor provided that the maximum of the valence band is at the gamma point as well as the minimum of the conduction band. Energy gap values are reported in Table 1.
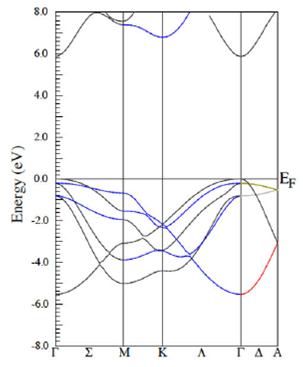
Figure 5 In this figure, it is displayed the AlN band structure in the wurtzite phase in the GGA approach.
The LDA calculations are not reported because they resemble those corresponding to the GGA approximation. It shall be mention that this phase also exhibits a direct gap for the electron transitions from the valence to conduction bands at the gamma point. Energy gap values are reported in Table I. It must be mentioned that the LDA calculations yield an energy gap in better agreement with the experimental value.
Recall that the AlN atomic structures in the hexagonal wurtzite phase each ion is bonded tetrahedrally to four neighbor ions of the other species. When projecting the orbitals that compose all bands, it is obtained complete s-p hybridization, which in turn induces covalent bonding. In the hybridization, the s orbital contributes in 1/4 and the p orbitals in 3/4 to yield the sp3 covalent bonds. Similar situations take place in the zinc blend structure.
The band structure of the AlN in the zinc blende phase is reported in Figure 6. Similar to Fig. 5, the GGA approach has been used. Results exhibit indirect electron transitions from the valence band to the conduction band provided that the valence band maximum is at the gamma point while the conduction band minimum is at the X point. Similar features are obtained in the LDA approach. It is noted that the energy gap is quite similar in both theories, with the value being smaller than 6 eV.
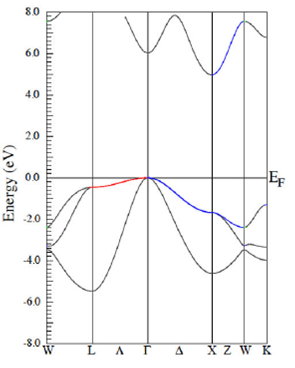
Figure 6 The figure displays the AlN band structures calculated within the GGA approach, in the zinc blende structure.
In the zinc blende phase, each ion is bonded tetrahedrally to four neighbor ions of the other species. When projecting the orbitals that compose all bands, it is obtained complete s-p hybridization, which in turn induces covalent bonding. In the hybridization, the s orbital contributes in 1/4 and the p orbitals in 3/4 to yield the sp3 covalent bonds. This is similar to the wurtzite phase.
The band structure of the AlN in the rock salt (NaCl) phase is reported in Fig. 7. The GGA approach is taken into account. Results exhibit indirect electron transitions from the valence band to the conduction band; the valence band maximum is at the gamma point while the conduction band minimum is at the X point. The LDA approach yields a similar band structure, with results are not reported. It is noted that the energy gap in the GGA approach is greater than that of the LDA calculation. The former case is greater than 6 eV but in the latter case, the gap is smaller than 6 eV.
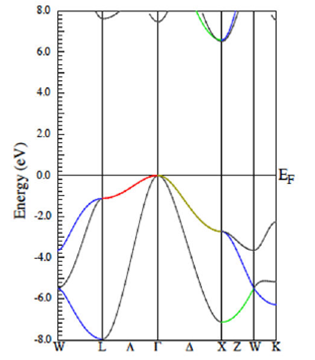
Figure 7 The figure displays the AlN band structures calculated within the GGA approach, for the rock salt phase.
In the rock salt phase, each ion is bonded to eight neighbor ions of the other species. Charge distribution plots indicate that electrons are mainly distributed at vicinities of either Al or N ions (calculations are not reported). When projecting the orbitals that compose all bands, it is obtained complete s-p hybridization. The projected density of states plots, as discussed below also corroborates this.
Note that the wurtzite phase yields a semiconductor with direct band transitions for electrons. In contrast, the cubic zinc blende and rock salt display indirect transitions. Let us remark that the energy gap calculated with Becke-Johnson potential meets quite well the experimental data.
4.Density of states
Aluminum nitride total density of states (DOS) and projected density of states (PDOS), within both approaches GGA and LDA are determined. In all figures, the Fermi level is the reference energy. Figure 8 displays the wurtzite total DOS (Fig. 8a) and PDOS (Fig. 8b) and Fig. 8c)) in the GGA approach. Note that the DOS exhibits an energy gap, in agreement with the band structure. The DOS (Fig. 8a) structure is interpreted in terms of the PDOS (Fig. 8b) and 8c)). At low energies, below the Fermi level, the contribution to the DOS is given by the Al-s and p orbitals, as well as the N-p orbital also contributes significantly. At energies above the gap, the largest contribution to the DOS is given by the Al-p and s, and the N-p orbitals.
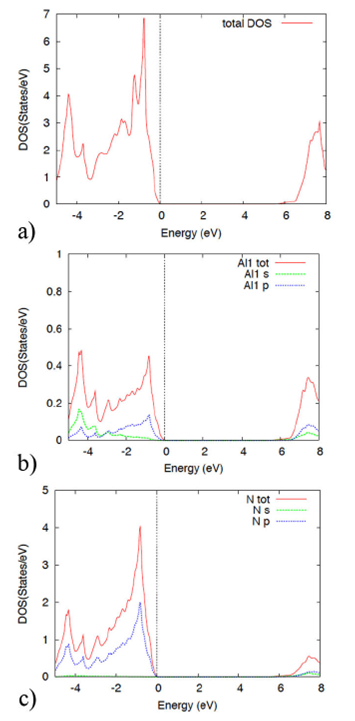
Figure 8 The figure displays the AlN density of states a) and the projected density of states b) and c) within the GGA theory, in the wurtzite structure.
Results of the total DOS and PDOS in the wurtzite phase within the LDA approach are not reported. It is worthwhile remarking that the DOS exhibits an energy gap in agreement with the band structure.
Figure 9 depicts the AlN total DOS and PDOS in the zinc blende phase calculated within the GGA. Note an energy gap in the DOS as in the band structure. The DOS feature in terms of the PDOS is as follows. At low energies, below the Fermi level, the most important contribution to the DOS is given by the Al-s and p orbitals. Note that the N-p orbital also contributes significantly. At energies above the gap, in the conduction band, the largest contribution to the DOS is given by the Al-s and p orbitals.
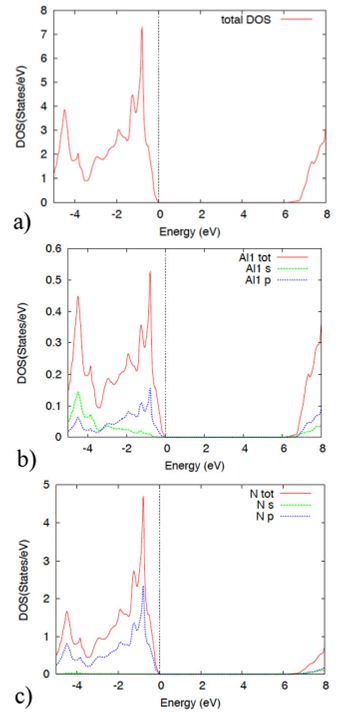
Figure 9 The figure displays the AlN density of states and the projected density of states within the GGA theory, in the zinc blende structure.
The total DOS and PDOS in zinc blende structure within the LDA approach have been calculated, however, they are not reported. In this approach, the DOS exhibits an energy gap, in agreement with the band structure.
Figure 10 display the AlN DOS and PDOS in the rock salt phase within the GGA approach. An energy gap is manifested as in the band structure. At low energies within the valence band, the most important contributions to the DOS are given by the Al-p, s, and N-p orbitals. At energies above the gap, within the conduction band, all s and p orbitals of Al and N contribute to the DOS.
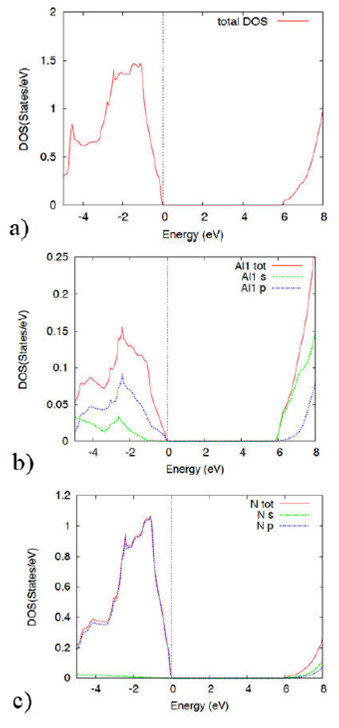
Figure 10 The figure displays the AlN density of states and projected density of states within the GGA theory, in the rock salt (NaCl) phase.
Finally, concerning the DOS and PDOS in the rock salt geometry within the LDA approach, calculations have been done, but they are not reported. It has been found that the DOS exhibits an energy gap as in the band structure.
5.Conclusions
Density functional theory calculations were performed to investigate the AlN structural phase transitions and electronic properties. Rock salt, zinc blende, and wurtzite structures have been considered for the calculations. The lattice parameters have been determined from the cohesion energy minima within both GGA and LDA approaches, results compare well with those reported. To improve the calculated energy gap value, the Becke-Johnson theory has been applied with results giving reasonable good agreement with the experimental data. The wurtzite phase shows band structure with direct electron transitions at the gamma point. However, the metastable zinc blende and rock salt phases exhibit indirect transitions. The wurtzite and zinc-blende structures exhibit sp3 hybridization, which in turn yield covalent bonds. The total density of states feature is described in terms of the projected density of states. To estimate the pressure at the structural phase transition the formation enthalpy is calculated. Results indicate that to achieve transitions small pressures are needed. The pressure needed to achieve a transition from wurtzite to rock salt is higher than that needed for the wurtzite to zinc blende.

