











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1.Introduction
The molecules of polyatomic gases are considered to have internal degrees of freedom that are related to internal (rotational, vibrational, and electronic) energy states. At very low temperatures, the degrees of freedom are frozen. By increasing the temperature, the rotational modes are excited. The vibrational modes are excited at higher temperatures, where, normally, the rotational modes are completely excited. The electronic modes are excited at the very highest temperatures. A rational thermodynamic theory for a diatomic gas with one excited mode was developed at first by Miiller and an extended thermodynamic theory by Kremer. Their approach is based on the equations of the balance of mass, momentum, and internal energy customary in thermodynamics and is supplemented by a general equation of balance for the vibrational energy. These equations of balance and simple constitutive equations give rise to field equations which a thermodynamic process must satisfy 1-5.
There has been an increasing interest in the study of the partition function and its derivatives of diatomic molecules because it is the essential link between the coordinates of microscopic systems and the thermodynamic properties. In the atomic and molecular physics, the interaction between atoms in diatomic and even in polyatomic molecules is usually described by the Morse potential. The Morse potential gives an excellent description of the interaction between the two atoms in a diatomic molecule. The several studies show that it yields an exactly solvent Schrödinger problem. Besides this potential is the most simple and realistic anharmonic potential model, which has been widely used in the description of the vibrational motion of diatomic molecules. Finally, the exact solutions of the Schrodinger equation with Morse potential based on the Pekeris approximation have been obtained 6,7: In the last decade a large community of researchers has been involved a search of approximate solutions for wave equations (non-relativistic or relativistic) including the centrifugal term and subject to different potential functions V(r). The main characteristic of these solutions lies in the substitution of the centrifugal term by an approximation so that one can obtain an equation, normally hypergeometric, which is solvable. Pekeris is the pioneer in the study. He managed to obtain analytic solutions for the radial Schrodinger equation with the Morse potential , through expansion for the centrifugal term.
Taking advantage of the exact energy spectrum of the Morse potential, we could analytically obtain the individual partition function, and consequently the thermodynamics functions can be deduced: analytical representations of thermodynamic functions of gases over the whole temperature range from zero to the thermal dissociation limit have aroused much interest in dealing with diatomic and polyatomic systems. Through the exact form of its spectrum of energy, the vibration partition function, which is of great importance to many issues in chemical physics and engineering, can be obtained. Following its definition, the molecular vibrational partition function can be calculated by direct summation over all possible vibrational energy levels. Many efforts have been made to acquire explicit expressions of partition function for molecular potential energy models in diatomic and polyatomic molecules: in this context, and also in the non-relativistic case, the investigation of thermodynamic functions of some type of potentials such as Morse and improved Manning-Rosen potentials, improved Rosen-Morse and Tietz oscillators, through a partition function and its derivatives with respect to temperature, were an important field of research in the literature 9-14,18-25.
Strekalov 15,16, derived a simple analytical formula for the partition function of Morse oscillators. With the cumulant expansion method, the approximation equally suitable within the whole range of temperatures, where a molecule exists as a bound system, has been obtained. Based on the Poisson summation formula (see Appendix A) and in order to reduce the errors with the experimental results, he proposed an accurate closed-form expression for the partition function in the case of the Morse oscillator. His approach becomes a most useful method used to calculate the thermal properties of some diatomic molecules for a different type of potentials (see following Refs 9-14,18-25). Recently, in the same context, another approach has been proposed: based on the Euler-Maclaurin formula (see Appendix A), the author has studied the case of the one-dimensional Morse potential in its q-deformed version: the results found are promising.
Now, before to give the principal aims of our works, we are in obligation to make some remarks about the difference between both methods described above.
-
The Poisson summation is an equation that relates the Fourier series coefficients of the periodic summation of a function to values of the function’s continuous Fourier transform. For appropriate functions f, the Poisson summation formula may be stated as :
where
-
The Euler-Maclaurin formula provides a powerful connection between integrals and sums. It provides expressions for the difference between the sum and the integral in terms of the higher derivatives f evaluated at the endpoints of the interval. It has the following definition 17-19:
where B 2p are the Bernoulli numbers, f 2p−1 is the derivative of order (2p − 1). Both methods are basic examples of deeper ideas. But at their core, the underlying ideas are very different.
The Euler-Maclaurin formulas are used to give high accuracy estimates of integrals in standard numerical analysis methods, as discrete sums with understandable error terms are computable and estimable. Thus, this method is the most important method for the summation of infinite series. Also, this formula is a bit tedious to use, and the amount of work depends on the derivatives.
The Poisson summation is very different in nature. It concerns Fourier series, or more fundamentally, the Fourier transform. This looks superficially similar to the integrals in Euler-Maclaurin summation, but the underlying ideas are substantively different. So the Poisson formula usually does not give an immediate answer but is a transform allowing other procedures to be applied.
Thus, the purpose of the present work is to give a simple analytical expression of the partition function of the Morse potential via the well-known Euler-Maclaurin method. This approach also enables one to estimate the influence of rotation-vibration interaction effects on the thermal properties of diatomic molecules. Precisely, we obtain the basic thermodynamic functions in terms of the vibrational quantum number l: in to our best knowledge, the study of these quantities by varying the quantum numbers l and n does not exist in the literature. The paper is organized as follows. In Sec. 2, we review the eigensolutions of three-dimensional Morse potential. Section 3 is devoted to deriving the vibrational partition function of our problem in question in three dimensions: through this function, all basic thermodynamic functions, such as the partition function and vibrational specific heat, are obtained. Section 4 will be a conclusion.
2.The behavior of the eigenvalues of the Morse potential in three dimensions
The time-independent Schrodinger equation for an arbitrary potential V(r) is given by
with the total wave function and the three-dimensional Morse potential are written as follows:5,6
where D is dissociation energy, r e is the equilibrium internuclear distance and a is the parameter controlling the width of the potential well. In order to solve Eq. (3), we first put x= (r − r e )/r e and then rewrite the potential function in terms of x as follows
Now, inserting Eqs. (6) and (4) into (3) we obtain
where n and l are the vibration-rotation quantum numbers, μ is the reduced mass of the diatomic molecule, and E nl is the appropriate energy eigenvalue. Now adopting the following approximation 7,8
with
and substituting (8) into (7), equation (7) is transformed into
with
Solving Eq. (12), we found that the rotation-vibrational energy levels of the Morse potential for diatomic molecules are given by
with n max denotes the upper bound. In this stage, two remarks seem important to notice:
Equation (16) is obtained by using the Pekeris approximation. The validity of this approximation depends on the magnitude of the rotational quantum number l: so it is not reliable for higher values of l27.
The closest to the maximum value allowed can be determined by putting the following condition dE/dn= 0 : following this condition, we obtain that
with ζ and η are defined by Eqs. (14) and (15).
Now, we are ready (i) to compute the ro-vibrational energy levels of diatomic molecules and (ii) to discuss our results about the variation of the energies in different situations. For convenience, we choose four molecules, H2, LiH, HCl, and CO, which have been most widely studied in the literature. The spectroscopic parameters of selected molecules are shown in Table I .
TABLE I Spectroscopic parameters of selected molecules used in the present calculation.
| Molecule | μ(amu) | D(eV) | a(A-1) | re |
| HCl | 0.9801045 | 4.61907 | 2.38057 | 1.2746 |
| H2 | 0.50391 | 4.7446 | 1.440558 | 0.7416 |
| CO | 6.8606719 | 11.2256 | 2.59441 | 1.1283 |
| LiH | 0.8801221 | 2.515287 | 1.7998368 | 1.5956 |
In Fig. 1, representative plots are given, with respect to the vibrational quantum number l at four selected values of n= 0, 5, 10, 50 for H2, LiH, HCl and CO molecules. In the other hand, Fig. 2 correspondings to the energy changes for rotational quantum number n at four selected l values, namely 0, 10, 20, 30. Following these figures, some remarks can be made 28:
The choice of a fixed value of n is a consequence of the fact that this potential supports a limited number of bound states for these molecules; the estimated n max for our molecules under consideration are shown in Table II; these values are obtained by using that ℏc= 1973.269 eV, Å and 1amu = 931.5×106 eV (Å)−1. Following this table, contrary to the case of H2, the number of quantum levels are finite and decrease with the quantum number l.
These figures allow us to obtain the allowed values of l leaving our spectrum of energy negative (bound states): the positive values of energy mean that the state is in continuum.
Also, it is seen that the energies E nl versus n plots for different l become more closely spaced, with H2 showing maximum sparsity. The rate of increase in energy, in general, increases as one move toward H2→CO→HCl→LiH, showing almost a linear behavior for CO. On the other hand, E nl versus l plots for all four molecules remain well separated. For a given molecule, E nl versus l seems to change far less appreciably as n progresses to higher values. As a special case, we observe that the plots for HCl remain quite similar to those in LiH.
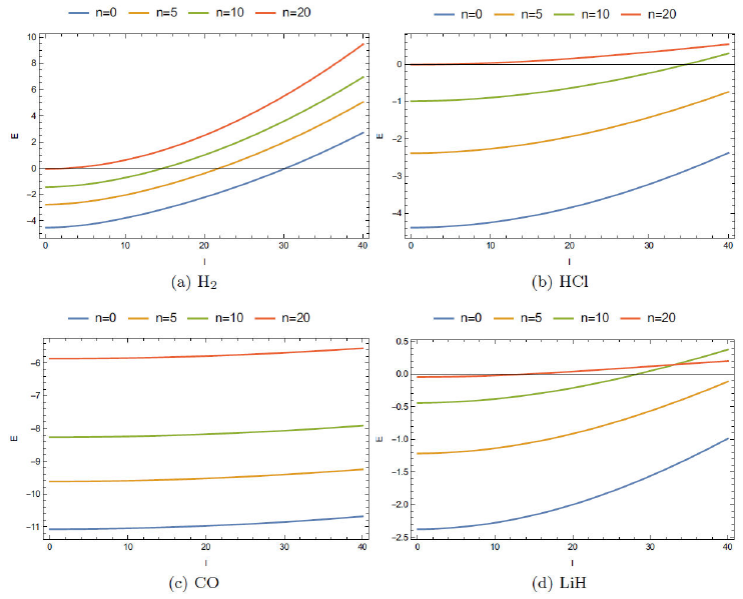
FIGURE 1 The ro-vibrational energy level to vibrational quantum number l for different values of vibrational quantum number n.
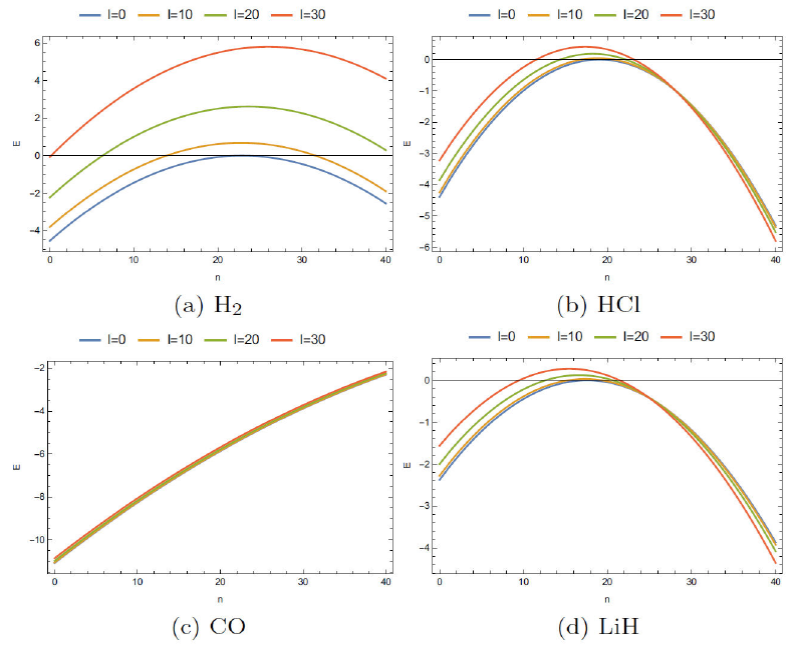
FIGURE 2 The ro-vibrational energy level versus vibrational quantum number n for different values of vibrational quantum number l.
TABLE II The values of nmax for H2, LiH, HCl and CO molecules.
| l | H2 | HCl | LiH | CO |
| 0 | 23 | 19 | 18 | 74 |
| 10 | 23 | 18 | 17 | 74 |
| 20 | 24 | 18 | 16 | 73 |
| 30 | 26 | 17 | 15 | 72 |
| 50 | 33 | 14 | 12 | 72 |
| 70 | 42 | 10 | 6 | 70 |
Finally, in order to understand well these observations, we have plotted the vibrational energy level vs both vibrational quantum numbers (n,l) in Figs. 3 and 4, following these figures, we observe that the molecule CO shows a linear behavior, and both molecules HCl and LiH have a similar variation. To our knowledge, such detailed energy plots have not been presented, and we hope these results would be helpful for future studies: precisely, to show the interval when the spectrum of energies becomes positive: this condition can be used as a rule of the selection of allowed values of l and n and consequently, used them for calculating the thermal properties of these molecules.

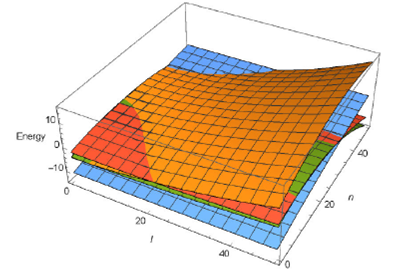
FIGURE 4 The vibrational energy level versus both vibrational quantum numbers (n; l) for four diatomic molecules H2, CO, HCl, LiH.
3.1Vibrational partition function
As we know, all thermodynamic quantities can be obtained from the partition function Z; therefore, the partition function of the system is the starting point to derive all thermal properties of the system in question. This function can be calculated by direct summation over all possible vibrational energy levels available to the system. Given the energy spectrum, the partition function Z at finite temperature T is obtained through the Boltzmann factor is
with β= 1/k B T, where k B is the Boltzmann constant.
Now in order to calculate the Z function, we use the Euler-MacLaurin formula: according to this approach, the sum transforms to the integral as follows (see Appendix A):
where B 2p are the Bernoulli numbers, and f 2p−1 is the derivative of order (2p−1). Up to p= 3, the partition function Z is written as
with B 2 = 1/6 and B 4 = −1/30 and
with E nl is given by Eq. (16). In what follows, all thermodynamic properties of the three-dimensional Morse potential, such as the free energy, the entropy, total energy, and the specific heat, can be obtained through the numerical partition function Z. These thermodynamic functions for the diatomic molecules system can be calculated from the following expressions:
In our case, we focus only on the study on the partition function Z and the specific heat C v . The influence of the quantum parameter l on these functions will be also examined.
3.2 Applications for some diatomic molecules
In the present work, we choose four diatomic molecules: H2, HCl, LiH, and CO, which have been most widely studied in the literature. The typical values of molecular constants for the electronic state of these molecules are given in Table I.
With the aid of these values, the vibrational partition function Z is determinate: via this function, the thermal properties of these molecules can be found easily. These thermodynamic functions are represented versus the inverse of temperature β= 1/k B T and the quantum number l. The Fig. 4(d) show some thermal quantities for the following diatomic molecules, H2, HCl, LiH, and CO: from this figure, some remarks can be made:
The Fig. 4(a) shows both the partition function Z and the reduced specific heat Cv/k B of H2 versus β for different values of vibrational quantum number l: it reveals that for increasing inverse temperature β the partition function is decreased as well. The variation of the reduced specific heat versus β for various values of l in the interval 0 ≤β≤ 5 is shown in Fig. 5 (a) . We observe that the C v /k B first increases with the increasing β until it reaches the maximum value β C = 1/k B T C and then decreases with it: we can see that this maximum value, T c , decreases when l increases (see Table II).
The Fig. 5 (b) shows both vibrational partition function Z and the reduced specific heat Cv/k B of CO as a function of β for different values of vibrational quantum number l: from this figure, we observe that both the partition function and Cv/k B are independent of the parameter l: so, we can state that all states of this molecule are identical.
Finally, Figs. 5(c) and 5(d) show both the vibrational partition function Z and the reduced specific heat Cv/k B of HCl and LiH as a function of β for different values of the quantum number l: from these figures, we observe that the behavior of both molecules is identical. This comportment is because both molecules have similar spectroscopic parameters (see Table I).
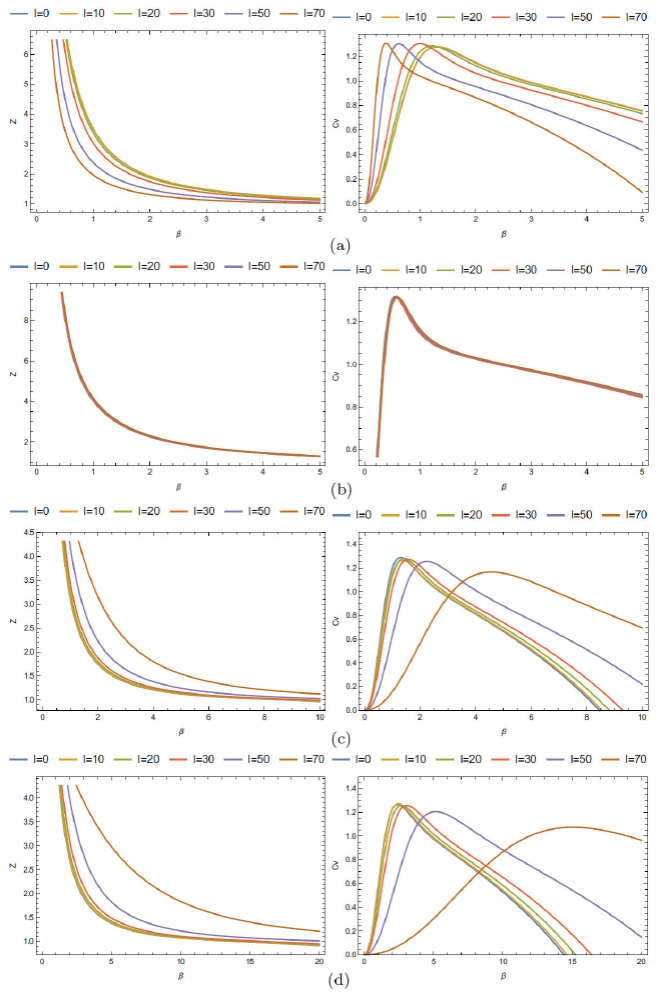
FIGURE 5 Thermal properties for the following Diatomic molecules, H2, H Cl, LiH, and CO. (a) Both the vibrational partition function Z and the reduced specific heat C=k B of H2 as a function of β for different values of vibrational quantum number l. (b) Both the vibrational partition function Z and the reduced specific heat C=k B of CO as a function of β for different values of vibrational quantum number l. (c) Both the vibrational partition function Z and the reduced specific heat C=k B of HCl as a function of β for different values of vibrational quantum number l. (d) Both the vibrational partition function Z and the reduced specific heat C=k B of LiH as a function of β for different values of vibrational quantum number l.
In Fig. 6 we show the behavior of the reduced specific heat of the diatomic molecules H2, HCl, LiH, and CO for two values of l: from this figure, we observe that the specific heat of H2, HCl is approximately the same, contrarily to the case of the molecules LiH and CO where the difference is well observed.
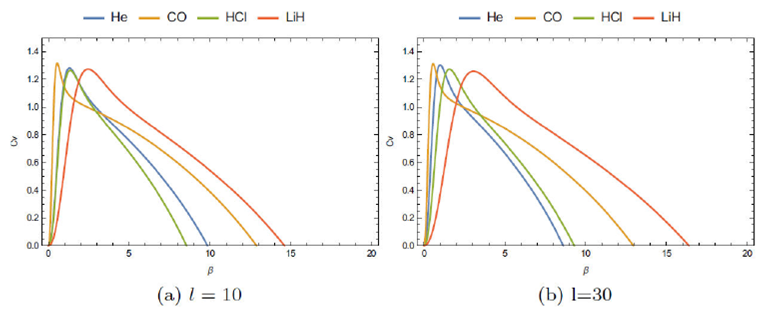
In Table III we show some values of the critical temperature T C = 1/k B β C , and its variation with the parameter l: as explained above, the condition of having the bound states (negative spectrum of energy) leads to the limitation of the quantum number l: following this, the relative value T C for each molecule, is T C H2 = 8788 (l≤10), T C HCl = 9355, 8923, 8923 K forl= 0, 10, 20 (l≤20), T C LiH = 5273, 4677 K for l= 0, 20 (l≤20) and T C CO = 20715 K. The case of the molecule CO requires some attention: all curves are identical and independent of the quantum number l. It means that all states of the molecule CO can be considered as in the S state (l= 0).
TABLE III The values of the critical temperatures TC (K) for the four diatomic molecules.
| l | H2 | CO | HCl | LiH |
| 0 | 8788 | 20715 | 9355 | 5273 |
| 10 | 8788 | 20715 | 8923 | 5273 |
| 20 | 9667 | 20715 | 8923 | 4677 |
| 30 | 11600 | 20715 | 7582 | 3866 |
| 50 | 18711 | 20715 | 5202 | 2265 |
| 70 | 31353 | 20715 | 2521 | 773 |
Also, we can see that the reduced vibrational specific heat C v /k B is more sensitive to n max than the vibrational partition function. The reason of this is twofold: (i) the specific heat depends upon the second derivative of the partition function and (ii) the expansion of the specific heat as a function of l is very clear in the figure for the different types of molecules: this enlargement can be explained by the decreases in the number of energy levels n max when l decreases except to the case of H2. Thus, for a system composed of diatomic molecules, a critical temperature value T C appears. This temperature means that the system becomes saturated and can no longer absorb more energy because all its excited states are occupied. We note here that this temperature was at first mentioned by . It corresponds to the maximum in the specific heat curves. These curves show the anomalous behavior of the specific heat when diatomic systems interact under the Morse potential.
Finally, the specific heat is an important physical quantity for testing the existence of a transition phase, as well as its nature (first or second-order). Recently, predictions of entropy for diatomic molecules and gaseous substances have been the subject of two recent studies . The authors have been obtained the exact expression of the vibrational entropy for (i) the improved Tietz oscillator and (ii) the Morse and Manning-Rosen oscillators. According to the results obtained by these authors, two remarks can be made: firstly, the diatomic molecules are treated as rigid rotors, and the interaction between two molecules is neglect. On the other hand, the specific heat values derived from experimental measurements are a combination of the translational, rotational, and vibrational specific heat. Thus, in order to compare our theoretical vibrational specific heat, it must have the experimental data of the total specific heat at first. But, we stress, though, as far as we know, we have not any data concerning the experimental values of the total specific heat that helps us to make a comparison with our theoretical results.
4.Conclusion
In this work, by using the vibrational energies obtained in the three-dimensional Morse potential, we have carried out a calculation of the vibrational partition function of the Morse potential for some diatomic molecules via the Euler-Maclaurin approach. Via this function, we have derived explicit expressions for the thermodynamic functions such as specific heat C v . We have analyzed the behavior of the specific heat: this analysis shows the existence of a critical temperature T C in the curves of the specific heat: this temperature decreases when l increases.

