











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink1. Introduction
Cancer is a group of more than 100 diseases that are formed by the uncontrolled proliferation of cells in various parts of our body. Untreated cases may result in serious discomfort or even death. The uncontrolled and rapid proliferation of cancer cells is due to the fact that the activity or number of growth factor receptors is excessively high. Among these factors, vascular endothelial growth factor (VEGFR-2) is a transmembrane receptor that plays an important role in endothelial cell development [1,2] and is thought to mediate the key effects of the endothelial-specific mitogen VEGF on cell proliferation and permeability. Therefore, the majority of VEGFR-2 actions are related to angiogenesis, which is one of the critical processes that affect growth and development of cancerous cells [3,4]. Blocking VEGFR-2 signalling pathway has become an attractive approach for the treatment of cancer [5,6]. Vatalanib (PTK787/ZK-222584), Semaxanib (SU5416), Sorafenib (BAY 43-9006), Vandetanib (ZD6474), Sunitinib (SU11248) are some of the antiangiogenic molecules that inhibit all vascular endothelial growth factor receptors. As the pharmaceutical field continues to evolve, efforts are underway to find new and effective VEGFR-2 inhibitors and development of different, and more effective anti-cancer agents than the known drugs is very important. The thiazoles in this area are a little more foreground and in the field of medicinal chemistry, thiazoles are of great interest since they are highly biologically active compounds, including numerous natural products and pharmaceutical agents [7,8].
Antibiotics are drugs used in the treatment of bacterial infection diseases, and are very important in terms of human health. the discovery of antibiotics in the last century decreased the infectious disease dependent deaths in a great extent. However, the increase in antibacterial resistance among bacterial pathogens is a worldwide thread constraining the different potential antibacterials. In the majority of existing pathogenic bacteria, this resistance to drugs is constantly emerging. Therefore, it is very important to have alternative agents that can be used as new antibacterial agents to prevent this serious problem.
Molecular structure states almost everything about the molecular function, if desired in detail. For a candidate anticancer and antibacterial agent, stability, softness and hardness, electronic behaviour and bonding capacity have to be well known. DFT study B3LYP exchange correlation functional gives structural and spectroscopic informations about the molecule which can be bridged to the function. In this framework, the B3LYP (Becke’s three-parameter exact exchange functional (B3) combined with the gradient-corrected correlational functional of Lee, Yang, and Parr (LYP)) hybrid method and the HSEH1PBE (it is deciphered as the Heyd-Scuseria-Ernzerhof hybrid combined with Perdew, Burke, and Ernzerhof’s exchange and correlation functions), called the HSE06 approach basis sets, are often used as adequate basis sets in the density functional theory (DFT) as a quantum chemical calculation method [10-18].
Very recently, in our previous work, 2-ethoxythiazole (C
At the present work, the experimental FT-IR and UV-vis (in chloroform, ethanol and N,N-dimethylformamide solvents) spectral results, the molecular geometry, the simulated vibrational and UV-vis (in gase phase and chloroform solvent) spectra, the proton and carbon-13 NMR chemical shift values, HOMO-LUMO, NBO analyses and NLO properties were calculated using DFT/B3LYP and HSEH1PBE with LanL2DZ which stands for “Los Alamos National Laboratory 2-Double-Z” basis set. Furthermore based on the above consideration, we performed molecular docking studies into the active sites of VEGFR-2 kinase and KAS III.
2. Experimental
5-Acetyl-2,4-dimethylthiazole (99%, Alfa Aesar), chloroform (99%, Merck), ethanol (99.9%, Merck) and N,N-dimethylformamide (99.8%, Merck) used in this work were obtained from commercial sources, and were used without any purification. IR spectrum of the 5-acetyl-2,4-dimethylthiazole was recorded at room temperature on a Schimadzu IR Prestige-21 FT-IR (Fourier Transform Infrared) Spectrometer with a resolution of 4 cm
2.1. Computational details
All calculations were carried out using Gaussian 09 program package with the GaussView 05 molecular visualization program on personal computer [20,21]. The molecular structure and vibrational computations of 5-acetyl-2,4-dimethylthiazole molecule were calculated by using Becke-3-Lee Yang Parr (B3LYP) and HSEH1PBE density functional theory methods with LanL2DZ basis set in ground state, respectively. Since the other well-known basis set lead us to imaginary vibrational frequency values, the mentioned basis set was used, and so the optimized molecular structure of title molecule in the ground state was obtained at the B3LYP/ and HSEH1PBE/ LanL2DZ levels. By considering the well-known systematic errors which come from the negligence of anharmonicity, electron correlation and basis set deficiencies, and furthermore, the computation in DFT method was performed in gas phase of isolated molecule while the experimental measurements were taken in solid phase for the title molecule [22,23], the calculated vibrational wavenumbers were scaled as 0.961 for frequencies higher than 800 cm
2.2. Molecular Docking
Molecular docking studies of 5-acetyl-2,4-dimethylthiazole was performed by on SwissDock web server using EADock DSS algorithm [30]. High resolution crystal structure of vascular endothelial growth factor (VEGFR-2) (PDB ID: 2XIR) and
3. Results and Discussion
3.1. Molecular structure
The optimized molecular structures at the HSEH1PBE/Lan L2DZ levels basis set of the 5-acetyl-2, 4-dimethylthiazole compound are in Fig. 1. In Table I we only present some selected bond lengths, bond angles and dihedral angles which were calculated at the B3LYP and HSEH1BPE /LanL2DZ levels for the title molecule.
TABLE I The calculated bond lengths (Å), bond angles and dihedral angles (°) for 5-acetyl-2,4-dimethylthiazole at the B3LYP and HSEH1PBE/LanL2DZ levels.
| Parameters | B3LYP | HSEH1PBE |
|---|---|---|
| Bond lengths | ||
| C1-S4 | 1.8141 | 1.8001 |
| C1-N5 | 1.3203 | 1.3171 |
| C1-C11 | 1.4991 | 1.4906 |
| C2-C3 | 1.3937 | 1.3893 |
| C2-N5 | 1.4025 | 1.3952 |
| C2-C6 | 1.5094 | 1.5005 |
| C3-S4 | 1.8203 | 1.8060 |
| C3-C10 | 1.4755 | 1.4685 |
| C10-C15 | 1.5227 | 1.5126 |
| C10-O19 | 1.2588 | 1.2539 |
| Bond angles | ||
| S4-C1-N5 | 114.2158 | 114.3176 |
| S4-C1-C11 | 121.5027 | 121.4847 |
| N5-C1-C11 | 124.2814 | 124.1977 |
| C3-C2-N5 | 114.7115 | 114.614 |
| C3-C2-C6 | 128.6948 | 128.6239 |
| N5-C2-C6 | 116.5937 | 116.762 |
| C2-C3-S4 | 110.2424 | 110.3151 |
| C2-C3-C10 | 132.679 | 132.6023 |
| S4-C3-C10 | 117.0786 | 117.0827 |
| C1-S4-C3 | 87.3958 | 87.5172 |
| C1-N5-C2 | 113.4344 | 113.2361 |
| C3-C10-C15 | 119.8574 | 119.6661 |
| C3-C10-O19 | 119.9356 | 119.9199 |
| C15-C10-O19 | 120.207 | 120.414 |
| Dihedral angles | ||
| N5-C1-S4-C3 | 0.0013 | 0.0033 |
| C11-C1-S4-C3 | 179.9998 | 180.0011 |
| S4-C1-N5-C2 | -0.0008 | -0.0023 |
| C11-C1-N5-C2 | -179.9993 | -180.0 |
| N5-C2-C3-C10 | 180.0008 | -179.9982 |
| C6-C2-C3-S4 | 180.0004 | -179.9988 |
| C6-C2-C3-C10 | 0.0 | 0.0 |
| C3-C2-N5-C1 | -0.003 | -0.0005 |
| C6-C2-N5-C1 | 179.9989 | -179.999 |
| C2-C3-S4-C1 | -0.0014 | -0.0034 |
| C10-C3-S4-C1 | 179.9983 | 179.9976 |
| C2-C3-C10-C15 | 0.0017 | -0.0009 |
| C2-C3-C10-O19 | 179.9983 | -179.9976 |
| S4-C3-C10-C15 | 180.0012 | 179.9978 |
| S4-C3-C10-O19 | -0.0021 | 0.0012 |
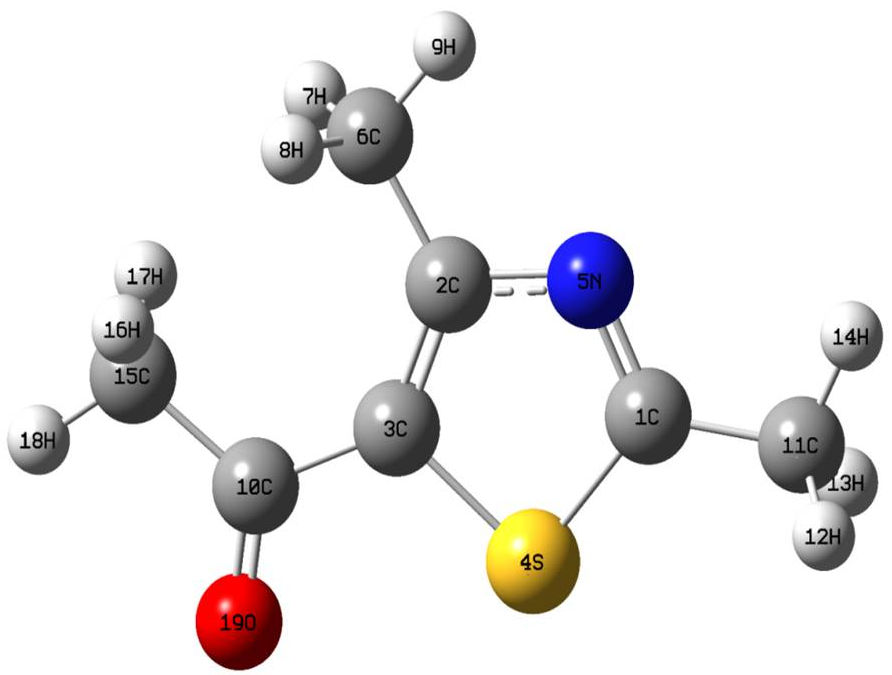
Figure 1 The optimized molecular geometry of 5-acetyl-2,4-dimethylthiazole at the B3LYP/LanL2DZ level.
By considering the Table I and Fig. 1, in thiazole ring the double bond C1=N5 and the single bond C2-5N were calculated as 1.3203/1.3171 and 1.4025/1.3952 Å at the B3LYP and HSEH1BPE /LanL2DZ levels, respectively. Likewise, the bond lengths C1-S4 and C3-S4 were found as 1.8141/1.8001 and 1.8203/1.806 Å at the mentioned levels, respectively, which are the longest bond lengths of the title molecule. On the other hand, in CH
3.2. Vibrational analysis
The experimental and simulated IR spectra of the 5-acetyl-2,4-dimethylthiazole compound (C
TABLE II Comparison of FT-IR and calculated vibration frequencies for the 5-acetyl-2,4-dimethylthiazole.
| Mode | Assignments via PED% at HSEH1PBE level | FT-IR | Scaled freq. [cm-1]a | |
|---|---|---|---|---|
| [cm-1] | B3LYP | HSEH1PBE | ||
| 1 | ν (CH) 97% | 3058.49 | 3089.61 | |
| 2 | ν(CH) 78% | 3052.72 | 3084.05 | |
| 3 | ν(CH) 98% | 3049.44 | 3083.68 | |
| 4 | ν(CH) 100% | 3017.11 | 3049.69 | |
| 5 | ν(CH) 100% | 2999.31 | 3008.94 | 3042.80 |
| 6 | ν(CH) 100% | 2964.59 | 3004.72 | 3039.31 |
| 7 | ν(CH) 79% | 2961.56 | 2938.89 | 2961.81 |
| 8 | ν(CH) 80% | 2935.95 | 2960.55 | |
| 9 | ν(CH) 100% | 2924.08 | 2932.77 | 2957.62 |
| 10 | ν(OC) 80% | 1670.31 | 1560.83 | 1595.54 |
| 11 | ν(NC) 13%+v (CC) 51% | 1510.28 | 1498.29 | 1528.25 |
| 12 | ν(NC) 45%+v (CC) 26% | 1475.42 | 1500.61 | |
| 13 | β(HCH) 60%+τ (HCCC) 14% | 1467.75 | 1473.16 | |
| 14 | β(HCH) 61% | 1454.79 | 1461.08 | |
| 15 | ν(NC) 35% +β (HCH) 19% | 1446.61 | 1444.87 | 1448.45 |
| 16 | β(HCH) 48%+τ (HCCC) 11% | 1443.66 | 1447.39 | |
| 17 |
ν(NC) 10% + |
1432.61 | 1441.51 | |
| 18 | β(HCH) 62% | 1426.83 | 1427.95 | |
| 19 | β(HCH) 87% | 1388.61 | 1392.94 | |
| 20 | β(HCH) 94% | 1386.64 | 1390.82 | |
| 21 | β(HCH) 86% | 1371.39 | 1366.00 | 1371.08 |
| 22 | ν(CC) 42% +β (CCN) 24% | 1317.38 | 1295.79 | 1326.23 |
| 23 | ν(NC) 37%+v (CC) 22% | 1269.52 | 1223.73 | 1252.40 |
| 24 | ν(CC) 27%+β (CNC) 12%+?(CCN) 12%+?(HCCS) 22% | 1182.38 | 1149.78 | 1170.30 |
| 25 | ν(CC) 12% +τ (HCCC) 20% | 1056.99 | 1044.35 | 1057.36 |
| 26 | τ(HCCC) 47%+γ (CCNC) 10% | 1041.58 | 1041.12 | 1046.04 |
| 27 | β(HCH) 20%+τ (HCCS) 58% | 1030.12 | 1034.66 | |
| 28 | ν(NC) 12%+τ (HCCC) 20% | 1021.25 | 1031.93 | |
| 29 | τ(HCCC) 45%+γ (OCCC) 20% | 1016.49 | 1016.58 | 1021.55 |
| 30 | v(NC) 10%+v (CC) 12%+τ(HCCC) 20% | 958.62 | 976.74 | 991.25 |
| 31 | ν(NC) 18%+ v(CC) 11% | 931.62 | 925.18 | 938.16 |
| 32 |
ν(CC) 41%+β (CCN) 12%+ |
860.45 | 885.11 | |
| 33 | τ(HCCC) 18%+τ (CCNC) 30%+ |
686.66 | 691.43 | 698.30 |
| 34 |
ν(SC) 21% +v (CC) 12%+ β (CCN) 15%+ |
667.37 | 667.11 | 678.55 |
| 35 | ν(SC) 39% + β (CCN) 11%+β (SCN) 12% | 636.74 | 656.33 | |
| 36 | τ(HCCC) 20%+τ (SCNC) 15%+γ (OCCC) 31% | 618.39 | 624.61 | |
| 37 | β(OCC) 36%+ β (CNC) 21% | 598.29 | 579.54 | 588.89 |
| 38 | ν(CC) 21%+v (SC) 18% +β (OCC) 15%+β (CNC) 12% | 567.07 | 558.12 | 570.49 |
| 39 | τ(SCNC) 32%+γ (OCCC) 12%+ γ(CSNC) 19% | 547.78 | 512.61 | 519.01 |
| 40 | β(CCC) 17%+β (SCN) 43% | 453.27 | 474.23 | 483.09 |
| 41 | ν(CC) 12%+β (CCN) 47% | 352.27 | 355.78 | |
| 42 | β(CCN) 17%+ β (OCC) 13%+ β (CCC) 40% | 327.45 | 329.70 | |
| 43 | τ(SCNC) 11%+γ (CSNC) 22%+ γ (CCSC) 36% | 296.21 | 299.09 | |
| 44 | β(CNC) 10%+ β (CCN) 74% | 282.40 | 284.30 | |
| 45 | τ(HCCC) 25%+ |
219.32 | 227.18 | |
| 46 | τ(CCNC) 27%+ γ(CCNC) 41% | 195.99 | 199.98 | |
| 47 | β(CCS) 65%+ β(CCC) 15% | 182.61 | 182.08 | |
| 48 | τ(CCNC) 16%+τ (SCNC) 24%+?(CSNC) 11%+ ?(CCSC) 31% | 112.89 | 114.09 | |
| 49 | τ(HCCC) 15%+τ (CCCC) 55% | 68.63 | 69.90 | |
| 50 | τ(HCCS) 84%+γ (CSNC) 12% | 49.29 | 44.26 | |
| 51 | β(HCC) 26%+τ (HCCC) 22%+τ (CCCC) 28% | 46.07 | 41.06 | |
ν: Stretching; β: in plane bending;
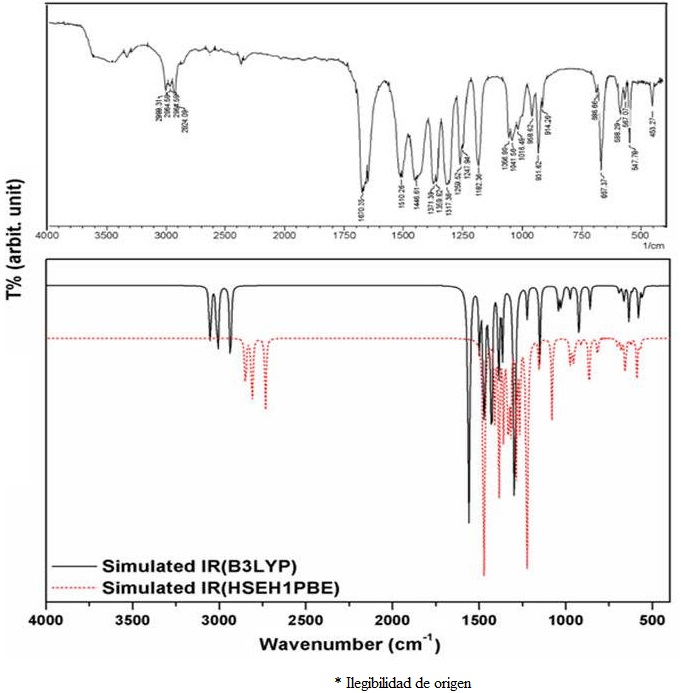
By considering Table II and Fig. 2, the
As for the v(C=N) stretching modes of thiazole ring of the title molecule observed at 1510.28 cm-1 was calculated as 1498.29/1528.25 cm-1 at the B3LYP and HSEH1PBE /LanL2DZ levels respectively, which are coupled by v(C-C) stretching vibration. Furthermore, the v(C-N) stretching modes observed at 1446.61 and 1269.52 cm-1 and calculated at the mentioned levels as 1444.87/1448.45 and 1223.73/1252.40 cm-1, respectively. On the other hand, the v(S-C) stretching mode is observed at 667.37 cm-1 as a strong band in Fig. 2 and calculated as 667.11/678.55 cm-1 at the mentioned levels, respectively.
3.3. NMR analysis
The nuclear magnetic resonance (NMR) spectroscopy is a powerful tool for determining the composition, structure and function of complex molecules and to compute the reliable magnetic properties which provide the accurate predictions of molecular geometries and the isotropic chemical shift analysis [37,38]. In this framework, the 1H and 13C NMR chemical shift values were calculated at the mentioned levels by using GIAO model. The calculated 1 H and 13 C NMR chemical shift values of the title molecule in gas phase and in the chloroform solvent at the B3LYP and HSEH1PBE /LanL2DZ levels are presented in Table III.
TABLE III The calculated 1H and 13C NMR chemical shifts (ppm)at the B3LYP and HSEH1PBE/LanL2DZ levels for 5-acetyl-2,4-dimethylthiazole.
| Atom | B3LYP | HSEH1PBE | ||
|---|---|---|---|---|
| Gas | Chloroform | Gas | Chloroform | |
| 1H | ||||
| H7 | 1.3815 | 1.5944 | 1.4394 | 1.6537 |
| H8 | 1.3814 | 1.5947 | 1.4394 | 1.6539 |
| H9 | 2.2407 | 2.1332 | 2.3638 | 2.2579 |
| H12 | 1.5738 | 1.7628 | 1.6497 | 1.8438 |
| H13 | 1.5737 | 1.7628 | 1.6496 | 1.8438 |
| H14 | 1.6947 | 1.6557 | 1.8062 | 1.7659 |
| H16 | 1.5028 | 1.7462 | 1.5678 | 1.8121 |
| H17 | 1.5029 | 1.7464 | 1.5678 | 1.8122 |
| H18 | 1.6591 | 1.612 | 1.7869 | 1.7451 |
| 13C | ||||
| C1 | 200.949 | 187.451 | 177.924 | 181.027 |
| C2 | 163.942 | 150.971 | 142.059 | 145.673 |
| C3 | 170.241 | 152.296 | 146.952 | 145.706 |
| C6 | 31.2081 | 14.7945 | 10.3229 | 10.490 |
| C10 | 206.325 | 194.838 | 185.19 | 190.306 |
| C11 | 31.9947 | 15.5823 | 11.0375 | 11.2336 |
| C15 | 42.5408 | 26.3812 | 21.5967 | 22.0522 |
By considering Table III, the outstanding point for the calculated
On the other hand, as seen in Table III, the highest calculated carbon-13 NMR chemical shift value in CHCl3 solvent of the title molecule at the B3LYP and HSEH1PBE /LanL2DZ levels was found as 194.838/190.306 ppm for the C10 atom which is connected to the O19 atom with high electronegativity while the lowest two calculated values in the chloroform solvent were found as 14.7945/10.49 ppm and 15.5823/11.2336 ppm for the C6 and C11 atoms, respectively.
3.4. UV-vis absorption and FMOs analysis
The experimental UV-vis in ethanol (EtOH), chloroform (CHCl3) and N, N-dimethylformamide (DMF) solvents and the simulated at the B3LYP and HSEH1PBE/LanL2DZ levels in gas phase and chloroform solvent spectra of the 5-acetyl-2,4-dimethylthiazole molecule are presented in Fig. 3. Furthermore, the maximum absorption wavelengths (λmax), excitation energies and oscillator strengths calculated by using TD-DFT/ B3LYP and HSEH1PBE/LanL2DZ levels of the title molecule in both gas and chloroform solvent are presented in Table IV.
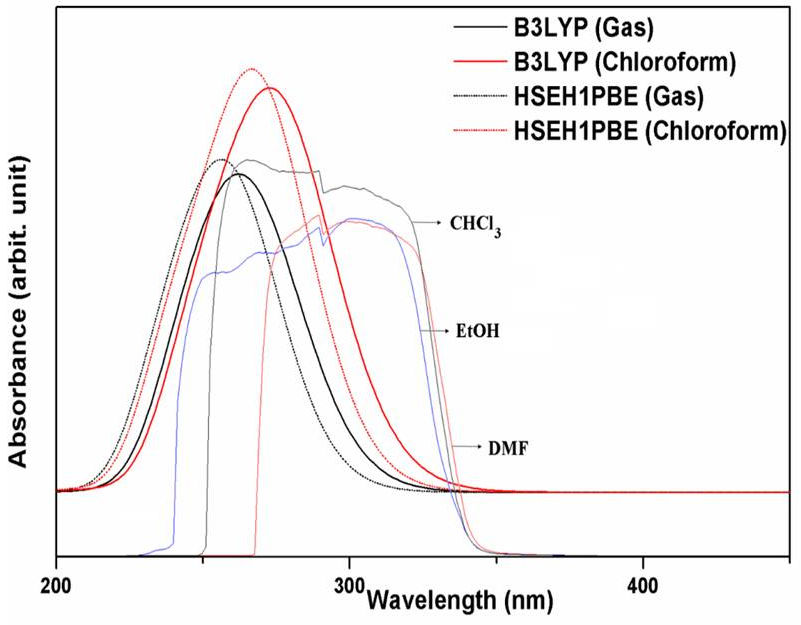
Figure 3 The experimental (in CHCl3, EtOH and DMF) solvents and simulated UV-vis spectra at the B3LYP and HSEH1PBE/LanL2DZ levels.
TABLE IV The experimental (in chloroform solvent) and calculated electronic transitions and oscillator strengths of 5-acetyl-2,4-dimethylthiazole at the B3LYP and HSEH1PBE/LanL2DZ levels.
| Transition | Experimental | B3LYP | HSEH1PBE | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chloroform | Gas | Chloroform | Gas | Chloroform | |||||||||
| λ (cm) | λ (cm) | E (eV) | Osc. | λ (cm) | E (eV) | Osc. | λ (cm) | E (eV) | Osc. | λ (cm) | E (eV) | Osc. | |
| 370.15 | 3.3495 | 0.0000 | 360.07 | 3.4434 | 0.0001 | 361.36 | 3.4310 | 0.0001 | 352.88 | 3.5288 | 0.0001 | ||
|
|
278 | 268.95 | 4.6099 | 0.1865 | 277.46 | 4.4685 | 0.2667 | 261.20 | 4.7468 | 0.2107 | 269.74 | 4.5964 | 0.2929 |
|
|
265 | 247.46 | 5.0103 | 0.1183 | 251.02 | 4.9391 | 0.1360 | 239.41 | 5.1788 | 0.1117 | 242.69 | 5.1087 | 0.1312 |
|
|
243 | 217.83 | 5.6918 | 0.0014 | 214.33 | 5.7849 | 0.0020 | 210.96 | 5.8771 | 0.0016 | 207.75 | 5.9678 | 0.0022 |
| 205.32 | 6.0386 | 0.0003 | 205.91 | 6.0212 | 0.0004 | 198.89 | 6.2337 | 0.0002 | 199.47 | 6.2157 | 0.0003 | ||
The experimental maximum absorption wavelengths (
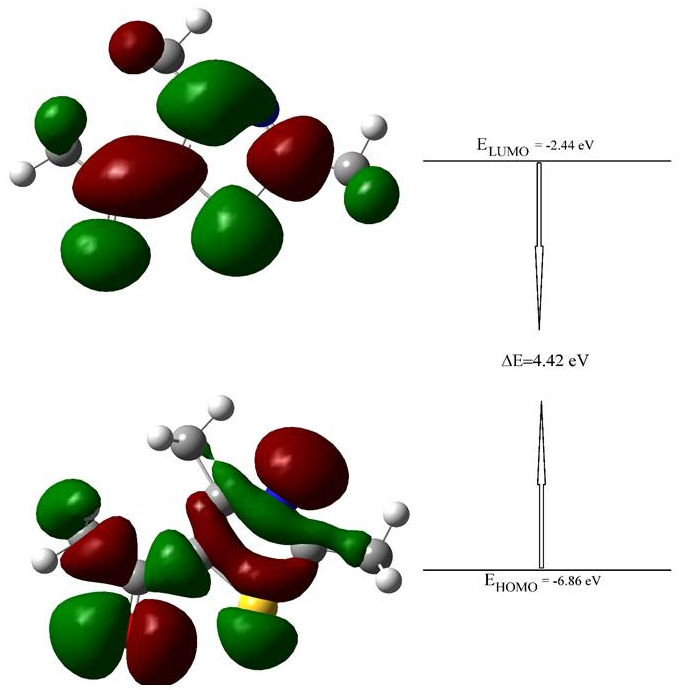
On the other hand, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) called the frontier molecule orbitals (FMOs) mean the outermost orbital filled by electrons and the first empty innermost orbital unfilled by electron, respectively. The HOMO is directly related to the ionization potential and behaves as an electron donor and the LUMO is directly related to the electron affinity and behaves as an electron acceptor. Therefore, the energy gap formed between HOMO and LUMO can be considered as a critical parameter and is an indicator in terms of the molecular chemical stability, and to identify the molecular electrical transport properties of any molecule. By using this energy gap, the molecular properties such as the chemical reactivity, polarizability, chemical hardness and softness, and electronegativity can be determined[40,41].
In this study, the 3-dimensional (3D) plots of the title molecule at the HSEH1PBE /LanL2DZ level are given in Fig. 4. The calculated energy gap value between the HOMO-LUMO for the title molecule is 4.42 eV at the mentioned level, as seen in Fig. 4. The large energy gap demonstrates the charge transfer occurs within of the title compound, and complex cannot be easily polarized. Besides, this value showing HOMO
3.5. NBO analysis
The NBO analysis provides an explanation of the intramolecular and intermolecular bondings and interactions among bonds in molecular system. Furthermore, it allows to study on the hyperconjugation interactions or charge transfers (ICT) in the molecules.
In this framework, a stabilizing donor-acceptor interaction is corresponded to the delocalization of electron density between occupied Lewis type orbitals and formally unoccupied non-Lewis orbitals. Therefore, in order to make clear the intra- and inter-molecular interactions in the molecular systems, the stabilization energies of the molecules have been studied by using second-order perturbation theory. For each donor NBO (i) and acceptor NBO (j), the stabilization energy E (2) associated with electron delocalization between donor and acceptor is estimated as [42,43]
where q i is the donor orbital occupancy, ε i and ε j are diagonal elements (orbital energies), and F ij is the off-diagonal NBO Fock matrix element (1). In this work, the results of second-order perturbation theory analysis of the Fock Matrix at the B3LYP and HSEH1PBE /LanL2DZ levels of theory for the 5-acetyl-2,4-dimethylthiazole molecule is presented in Table V.
TABLE V Second-order perturbation theory analysis of Fock matrix in NBO basic corresponding to the intramolecular bonds of 5-acetyl-2,4-dimethylthiazole calculated at the B3LYPand HSEH1PBE/LanL2DZ levels.
| Donor (i) | ED(i) (e) | Acceptor (j) | ED(j) (e) | E(2)a (kcal/mol) | E(j)-E(i)b (a.u.) | F(i,j)c (a.u.) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | HSEH1PBE | B3LYP | HSEH1PBE | B3LYP | HSEH1PBE | B3LYP | HSEH1PBE | B3LYP | HSEH1PBE | ||
|
|
1.96 | 1.96 |
|
0.02 | 0.02 | 7.01 | 7/.17 | 0.94 | 0.96 | 0.074 | 0.073 |
|
|
1.98 | 1.98 |
|
0.96 | 0.94 | 7.19 | 7.41 | 0.70 | 0.71 | 0.064 | 0.066 |
| LP1 (N5) | 1.89 | 1.89 |
|
0.04 | 0.04 | 8.46 | 8/.63 | 0.89 | 0.90 | 0.079 | 0.080 |
|
|
1.79 | 1.79 |
|
0.32 | 0.33 1 | 0.20 | 9.73 | 0.27 | 0.27 | 0.049 | 0.047 |
| LP2 (O12) | 1.90 | 1.91 |
|
0.06 | 0.06 | 15.61 | 15.79 | 0.69 | 0.70 | 0.094 | 0.095 |
| LP2 (S4) | 1.64 | 1.63 |
|
0.30 | 0.31 | 15.69 | 15.12 | 0.26 | 0.26 | 0.058 | 0.057 |
| LP2 (O19) | 1.90 | 1.91 |
|
0.05 | 0.05 | 17.26 | 17.42 | 0.61 | 0.62 | 0.093 | 0.094 |
| LP1(N5) | 1.89 | 1.89 |
|
0.09 | 0.09 | 17.43 | 1/7.15 | 0.54 | 0.55 | 0.087 | 0087 |
|
|
1.85 | 1.85 |
|
0.30 | 0.31 | 19.59 | 19/.38 | 0.34 | 0.34 | 0.076 | 0.076 |
|
|
1.79 | 1.79 |
|
0.18 | 0.18 | 24.10 | 23.62 | 0.28 | 0.28 | 0.074 | 0.073 |
| LP2 (S4) | 1.64 | 1.63 | π* (C1-N5) | 0.32 | 0.33 | 27.47 | 26.74 | 0.23 | 0.23 | 0.072 | 0.070 |
ED = electron density; aE(2) means energy of hyperconjugative interaction (stabilization energy);
b
Energy difference between donor and acceptor
By considering Table V, the stabilization energy values greater than 7.00 kcal mol-1 are listed. Therefore, the strongest stabilization energy values calculated at the B3LYP and HSEH1PBE/LanL2DZ levels were found as 27.47/26.74 kcal/mol, respectively and corresponds to the transition between the lone pair n electrons of S4 electrons and
3.6. NLO parameters
The calculated results of the molecular polarizabilities at the B3LYP and HSEH1PBE /LanL2DZ levels of the finite-field approach for the 5-acetyl-2,4-dimethylthiazole molecule are given in Table VI. Therefore, total static dipole moment
TABLA VI Total dipole moment (μ, in Debye), the mean polarizability (< α >, in 10-24 esu), the anisotropy of the polarizability (Δα, in 10-24 esu), the mean first-order hyperpolarizability (< β >, in 10-30 esu) for 5-Acetyl-2,4-dimethylthiazole calculated at the B3LYPand HSEH1PBE/LanL2DZ levels.
| Property | B3LYP | HSEH1PBE |
|---|---|---|
| μ | 3.10 | 3.09 |
| < α > | 14.46 | 14.31 |
| Δα | 10.45 | 10.13 |
| < β > | 3.38 | 3.32 |
| < β >* | 0.37289 |
* Taken from Ref. [44].
Therefore, the calculated values at the B3LYP and HSEH1PBE /LanL2DZ levels converted by using
NLO parameters of the molecular systems and in this work, the calculated
3.7. Molecular Docking
The binding properties of 5-acetyl-2,4-dimethylthiazole to the VEGFR-2 kinase (PDB ID: 2XIR) and acetoacetyl-ACP synthase (PDB ID: 1HNJ) proteins were investigated, and molecular modelling studies were performed for ligand-protein interactions. Molecular docking studies provide the best representation of the association of the studied compound with the receptor, as well as hydrogen bonds that play an important role in ligand-protein interaction which is required for inhibition activity. As a result of the molecular docking studies, it was found that the optimized structure of the 5-acetyl-2,4-dimethylthiazole binds to the protein by hydrogen bonding with certain amino acids in the binding region of both proteins. The obtained energy and hydrogen bond location informations are given in Table VII.
TABLE VII Full fitness score, binding energy and hydrogen bond information between the ligand-target molecule couples.
| Ligand-Target | Full fitnessscore (kcal/mol) | ΔG(kcal/mol) | H Bond Location(Length) |
|---|---|---|---|
| Ligand-2XIR | -1631.80 | -6.02 | N of ligand & |
| NH of Asp 1046 | |||
| (2.038 Å) | |||
| O of ligand & | |||
| Ligand-1HNJ | -1435.90 | -5.96 | NH of Leu 191 |
| (2.165 Å) |
According to the results, in the most stable docked pose between ligand-2XIR showed lowest interaction energy of -6.02 kcal/mol and was involved a hydrogen bonding interaction with Asp 1046 in the active site of 2XIR (Fig. 5). The amino acid Asp1046 had formed hydrogen bonding with nitrogen atom of 5-acetyl-2,4-dimethylthiazole (2.038 Å).
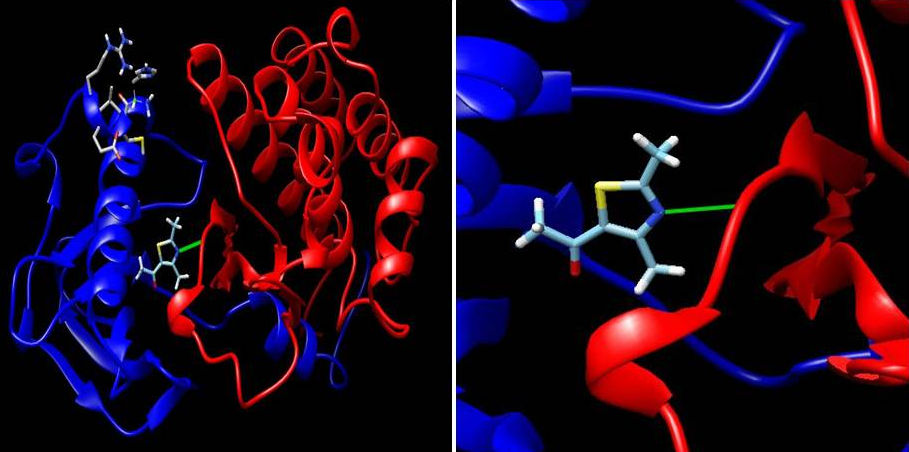
On the other hand, ligand-1HNJ interaction exhibited the higher free energy at -5.96 kcal/mol than the ligand-2XIR couple. 5-Acetyl-2,4-dimethylthiazole and 1HNJ interaction also exhibited a hydrogen bonding between O atom of ligand and NH of Leu 191 with a 2.165 Å bond length (Fig. 6). Since the ligand forms a shorter hydrogen bonding with the 2XIR, the bond between the two is much stronger than ligand-1HNJ couple. Full fitness score value of ligand-2XIR (Fig. 7) and ligand-1HNJ (Fig. 8) interactions were found to be as and -1435.90 kcal/mol, respectively. These scores were in accordance with the order of the binding energy values. Docking assay of 5-acetyl-2, 4-dimethylthiazole with 2XIR showed a better full fitness score value and more preferred interaction.
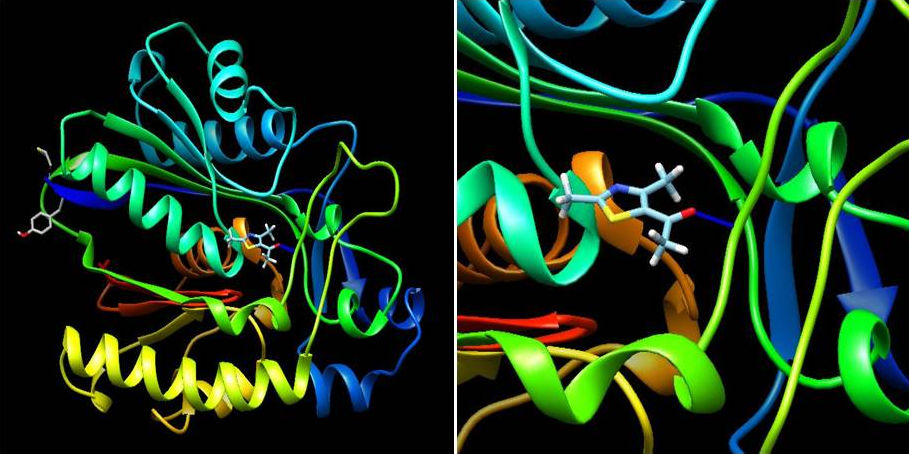
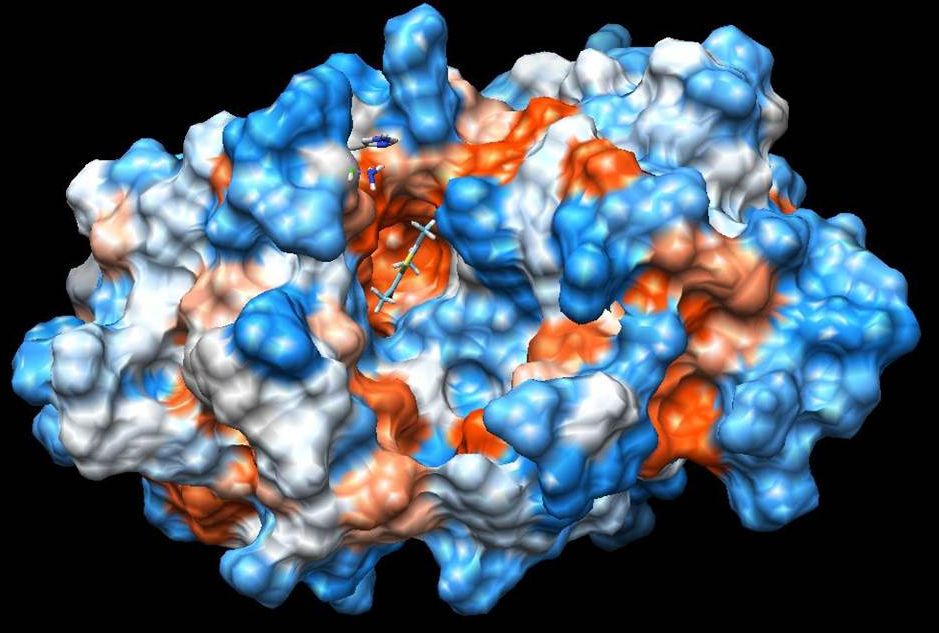
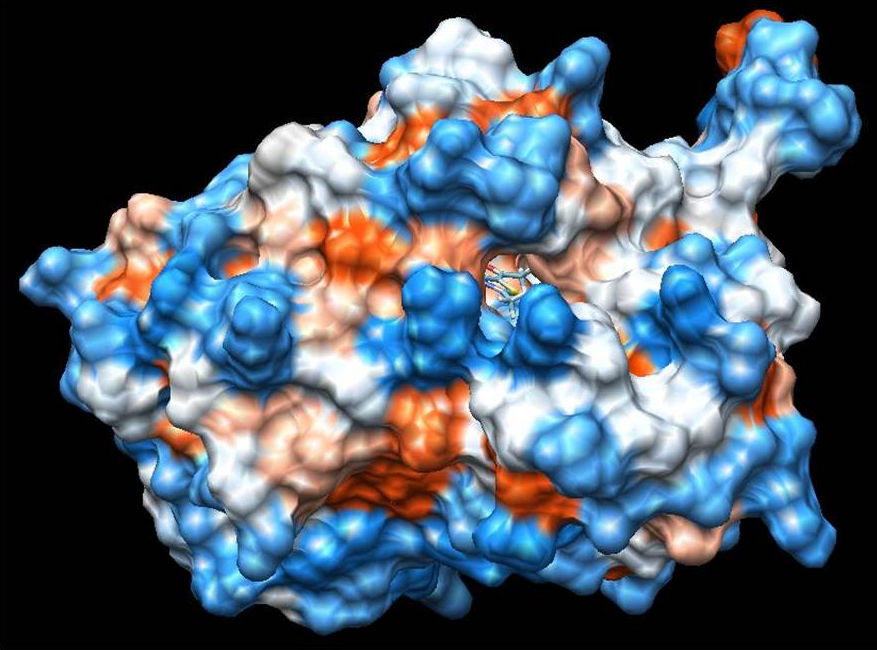
The docking study results clearly reveal that 5-acetyl-2,4-dimethylthiazole showed strong binding affinity towards the target proteins, achieving the best full fitness score and binding energy against 2XIR. However, full fitness score and binding energy for 1HNJ also appeared significant. On the basis of docking results, it was found that 5-acetyl-2,4-dimethylthiazole had potential to inhibit vascular endothelial growth factor receptor-2 kinase and
4. Conclusion
The present work indicates that the calculations on the 5-acetyl-2,4-dimethylthiazole molecule give us a reliable assignment for the NMR, IR and UV-vis spectra of the molecule. Meanwhile, the results of NBO analysis for title molecule exhibit that the calculated strongest stabilization energy values in

