











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink1. Introduction
The double perovskites A2BB’O6 systems (where A is an alkaline-earth, B and B’ are two different transition-metal elements), have become very attractive in view of their potential spintronic applications 1,2. In particular, the double perovskite Sr2FeMoO6 (SFMO) has a substantial low field magnetoresistance and combines half-metallic ferromagnetic character together with a high Curie temperature TC ∼ 420 K 3, thus giving the possibility of designing spintronics material operating at room temperature.
In the fully ordered double perovskite SFMO, FeO6 and MoO6 octahedron are alternating along the three crystallographic axes of the perovskite structure and Sr lies in the dodecahedral sites. Here, ferromagnetism and half-metallicity have been explained 4 from a strongly correlated picture, within this scheme Fe3+ (3d5) localized ions are in a highspin S = 5/2 configuration, the Mo5+ cores have S = 1/2 and there is one itinerant electron per formula unit (f.u.) which can hop to Fe sites only with an antiparallel orientation to the localized spin, stabilizing a ferromagnetic arrangement of local spins and fully opposite spin-polarized itinerant electrons. This formalism is consistent with ab initio calculations 3,5,6. The saturation magnetization is significantly lower than 4 μB per f.u. (Stotal = 5/2 − 1/2) expected from the polarized bands 3,7, a discrepancy has been attributed to some degree of disorder between Fe and Mo atoms which interchange their crystallographic positions, creating the antisites.
Increasing the number of conduction electrons in SFMO by substitution of divalent Sr2+ by trivalent La3+ ions in Sr2−y Lay FeMoO6 (SLFMO) seems to be a good mechanism to increases the Curie temperature (T C ) 8,9 because the increased number of electrons reinforces the ferromagnetic interactions between the Fe ions. However, this increase of TC cannot be accounted for by just increasing the carrier density, it has been suggested 4 that correlations within the conduction band could play a crucial role, giving rise by itself to a ferromagnetic instability.
Experimental results 9 have obtained an important increment of the Curie temperature, reaching 490 K for SrLaFeMoO6 (y = 1). The number of electrons per f.u. in SLFMO doped system is 1 + y while for the undoped SFMO compound is 1 electron per f.u. The substitution of Sr by La is associated with an increase of disorder in Fe and Mo ions, leading to a reduction of the magnetization, this fact is mainly for the increases of the magnetic moment on the Mo ions. The added electron occupies the minority spin band so that the total magnetic moment in the cell decreases by 1 μB. In this work, we present the Curie temperature behavior for the Sr 2−y La y FeMoO6 (0 ≤ y ≤ 1) doped system taking into account electronic correlation in Fe and Mo sites without disorder.
2. Model
In the ordered Sr2FeMoO6 system, Fe and Mo ions are contained in two interpenetrating sublattices α and β respectively, where the oxygen atoms bridge the Fe and Mo ions to form alternating FeO6 and MoO6 octahedra. The minority spin-down band of this half-metallic double perovskite consist of a strongly mixed Fe and Mo t 2g states crossing the Fermi level, while majority spin-up band, formed of t2g and eg states of Fe-d and Mo-d, shows a gap ∼ 0.5 − 0.8 eV. In our model we will use a tight-binding Hamiltonian considering only Fe and Mo sites, mainly because of the states near the Fermi energy come from Fe and Mo d-orbitals. We will also consider a strongly correlated picture of Fe3+ (d5) and Mo6+ (d0) states. In the ordered case, each electron on Fe3+ (α) site is surrounded by Mo6+ (β) ions and viceversa. Since the Fe ions have a strong Hund’s coupling, we take these orbitals as frozen with localized spin Si. Itinerant electrons from Mo move between Fe t 2g and Mo t2g orbitals via O-p states with the same symmetry, giving three degenerate two-dimensional bands with coordination z = 4 and a mixed valence configuration of Fe2.5+ and Mo5.5+. These itinerant electrons are coupled anti-ferromagnetically to the Fe localized spins, because all the five d-orbitals are occupied by an Fe electron with the same spin direction, it means, itinerant electron with spin σ can hop only to Fe sites with antiparallel localized spins (Pauli’s exclusion principle).
In Fig. 1 we show a scheme of Fe and Mo ions without disorder and considering only nearest-neighbors hopping. Fig. 1(a) shows the localized Fe (in squares) and itinerant Mo (in circles) spin configuration, while the Fig. 1(b) presents the itinerant electron that comes from the substitution of divalent Sr by trivalent La (pentagons) which goes to Mo sites. In both cases, itinerants electrons can hop to α lattice, it means, to Fe sites with spin down orientation.
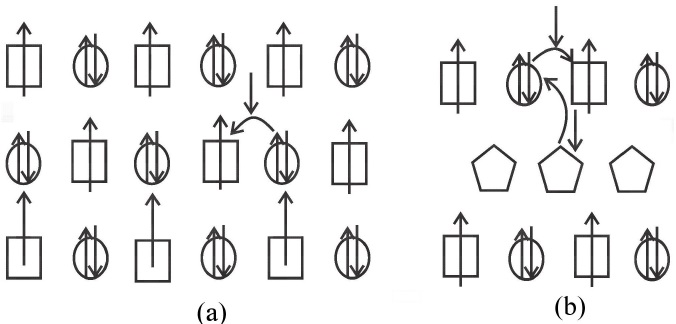
Figure 1 (a) Spin configuration for Fe (squares) and Mo (circles) ions in a perfect lattice, itinerant electron from Mo is shown too. (b) La (pentagons) ion provides an extra itinerant electron.
As we mentioned in the previous section, in view of studying TC, it is essential to take into account correlations both on Fe and Mo ions. Contrary to the assertion of Brey et al. 10, on Fe sites the inter-band is the only interaction intervening, it means, only one spin direction being possible on sites with + or − local spins. The intra-atomic correlations among itinerant electrons are given by
ν and ν′ label the three t2g orbitals and i, j correspond to sites occupied by Mo and Fe. Site energies are calculated using mean-field approximation to obtain the renormalized energies ẼFe,σ and ẼMo,σ, it means,
where ẼFe,↑
(ẼFe,↓) refers to Fe sites with
localized spin −(+).
and we used ⟨niνσ ⟩ = ⟨niσ ⟩/3 due to the degeneracy of the three t2g orbitals. We shall take
since these values have been shown to provide good results in disordered compound 11,12.
From the local Green’s functions within the renormalized perturbation expansion method 4,13, we calculate the density of states per orbital on Fe and Mo sites for itinerant electrons. In an alternating Bethe lattice in the limit of infinity coordination number, z → ∞, where zt 2 scales as w 2/4, w being half the bandwidth. The local average Green’s functions take the dynamical mean-field form
where εi is the corresponding on-site energy and the summation is over all nearest neighbors sites.
Expressions for the Green’s functions with an itinerant electron are given as: for spin ↓
and for spin ↑
where ẼFe,σ and ẼMo,σ denote the effective Fe and Mo site energies respectively including electronic correlation, we also introduce the probability ν ± = (1 ± m)/2 that an Fe ion has its localized spin + or −.
In the ordered state, the effective charge transfer energy in the ferromagnetic state for ↓ spin electrons is defined by ∆,
Here, EMo −EFe = ∆0, where ∆0 is the bare charge transfer energy for the non correlated case.
The charge transfer energy in SFMO is quite small 4, therefore in this paper we consider ∆ = 0 in order to reproduce the mixed-valence character in Fe2.5+ and Mo5.5+ observed experimentally 14,15. For ∆ = 0 in Eq. (9) and considering the values of the correlations parameters mentioned above, we obtain ∆0 = 2nw/3 for any concentration n. In particular, for n = 1 we have the SFMO compound and ∆0 = 2w/3.
The total density of states
where
where the number 3 is due to the degeneracy of the three t2g orbitals. The kinetic energy of the conduction electrons is determined by
To obtain the thermodynamical values of m(T) we need to calculate the minimum of the free energy
where S(m) is the entropy term of the local spins,
which is consistent with our approximation that these are either up or down. Finally, Curie temperature TC is calculated from m (TC) = 04.
3. Results and discussion
In Fig. 2 we show the density of states for the doping Sr 2−y La y FeMoO6 system, here the number of carriers increases by replacing divalent Sr2+ by trivalent La3+ as n=1+y (9). We keep ∆0=2w/3 as in pure Sr2FeMO6 and vary only n(y). Figure 2 also shows a Fermi energy represented by dashed line and the results of the density of states for y = 0, 0.5, 1.0. In the ferromagnetic case (m = 1) the system has a half-metallic character because electron adds occupies the minority spin band as we can see in Fig. 2(a), and also presents a gap in the middle of the band which reflects the fact that ∆ ≠ 0 due to the correlations although ∆ = 0 for y = 0 (n = 1). In Fig. 2(b) we observe how the density of states for the paramagnetic state (m = 0) goes to higher energies. In both cases, ferromagnetic and paramagnetic states, Fermi energy goes toward the gap.
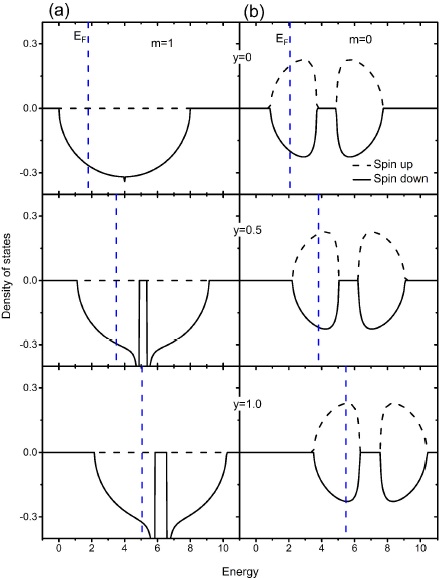
Figure 2 Density of states versus energy for (a) ferromagnetic (m = 1) and (b)
paramagnetic (m = 0) states as function of La
concentration (y). Dashed lines indicate the Fermi
energy (EF). electronic
correlation parameters are
In Fig. 3 we present the behavior of Curie temperature as function of La doping y for (0 ≤ y ≤ 1) as in the experimental results 9. Curie temperature increases as increasing La concentration reaching its highest value for y = 1, this values for ∆0 = 2w/3 (∆0 associated with Sr2FeMoO6) is by far too large as compared with experimental results. However, we have to remember that La doping induces disorder between Fe and Mo sites 9 which is detrimental to TC. This disorder is because the Fe atoms migrate to the Mo sites in order to balance the charge introduced by La doping into the cell, the number of oxygens atoms should be constant to keep the crystalline structure 16. Considering disorder, the variation of TC as function of y will certainly be slower than obtaining here. In our model, it is posible to introduce disorder 12 which will be very relevant on the calculation of the Curie temperature, unfortunately it is not yet known with certainty how disorder varies with La doping in these systems. Regarding the correlated (∆0 = 2w/3) and non-correlated (∆0 = 0) results we observe a substantial increase of TC due to the correlations effects on Fe and Mo sites.
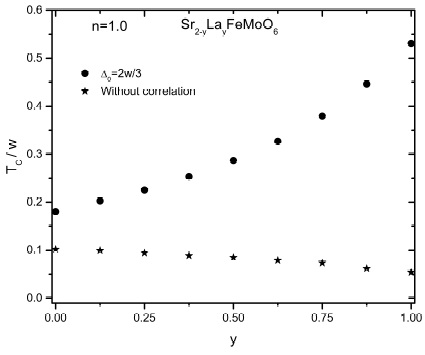
Figure 3 Curie temperature as function of La concentration (y) for Sr2−y Lay FeMoO6. The electronic correlation parameters are the same as those in Fig. 2.
In summary, substitution of Sr2+ by La3+ increases itinerant electrons in the Sr2−y Lay FeMoO6 compound which reinforces the ferromagnetic state and increases the TC. We have presented an electronic correlation study of this double perovskite compound within the dynamical meanfield approach. We obtained that our calculation of TC in Sr2−y Lay FeMoO6 for small La concentration, where disorder is not dominant and using ∆0 = 2w/3, is in qualitative agreement with experimental results.

