1. Introduction
It is well-known that electron paramagnetic resonance (EPR) technique is a powerful tool to study the environmental symmetry of the transition-metal ions doped into the crystals (or glasses) which is produced by the ligands around the metal ion and can also give valuable information about the electric fields of these impurity ions 1. The vanadyl ion (VO2+) with 3d1 configuration is probably the most stable among the molecular paramagnetic transition metal ions and is often used as an impurity probe for detecting the site symmetry of the central ion and the bonding nature with the ligands in the EPR studies 2,3 Due to the strong V4+-O2- covalence bonding in VO2+, many VO2+ complexes in crystals (or glasses) possess C
4v
symmetry with the axial symmetry for g and A values 4,5,6. For example, EPR investigations for VO2+ center in CaO-Al2O3-SiO2 glass-ceramic system were performed by Farah and the anisotropic g factors (g∥, g⊥) and the hyperfine structure constants (A∥, A⊥) were also obtained for this center 6. According to the EPR results, the oxygen octahedron around the central ion V4+ was found to suffer tetragonally compressed distortion 6. According to the EPR results, the oxygen octahedron around the central ion V4+ was found to suffer tetragonally compressed distortion 6. Based on the traditional crystal-field theory, theoretical calculations for EPR spectra of V4+ center in CaO-Al2O3-SiO2 were carried out through the complete diagonalization energy matrix method (CDM) and perturbation theory method (PTM) by Wei et al, 7 and the theoretical results show good agreement with the observed values (see Table I) 7. However, there may be some imperfections in their calculations. First, many adjusted parameters (i.e., the crystal field parameters B
20, B
40 and B
44, the orbital-reduction factor k and the core polarization constant κ) were introduced in their studies. Second, the contributions to EPR parameters from the ligand orbitals and SO coupling interactions were ignored for the strong covalent system CaO-Al2O3-SiO2: VO2+ characterized by the small orbital-reduction factor k(≈0.7≪1) 7. Since the SO coupling coefficient ζp(≈151 cm-14) of the ligand O2- is close to that (≈248 cm-11) of the central V4+ ion, these contributions from ζp and the p- (s-) orbitals of the ligands may be somewhat important for this center due to strong covalency. Finally, No information about the local structures has been determined for the octahedral cluster [VO6]8- in CaO-Al2O3-SiO2 system, since previous computations failed to correlate with the defect structures around the impurity. Due to the reason that analysis of EPR spectra can provide useful information about local defect structures of the cluster [VO6]8- in CaO- Al2O3-SiO2 system, and this would be helpful in understanding the properties of this glass-ceramic system. Thus, further studies on the above EPR parameters are of scientific and practical meanings. So, in this paper, the EPR parameters are quantitatively analyzed based on high-order perturbation formulas of these parameters for a d1 ion in tetragonally compressed octahedral, the contributions from the tetragonally compressed distortion as well as the SO coupling and the orbitals of the ligands are considered in these formulas.
Table I. The EPR parameters of the tetragonal VO2+ center in CaO-Al2O3-SiO2 system
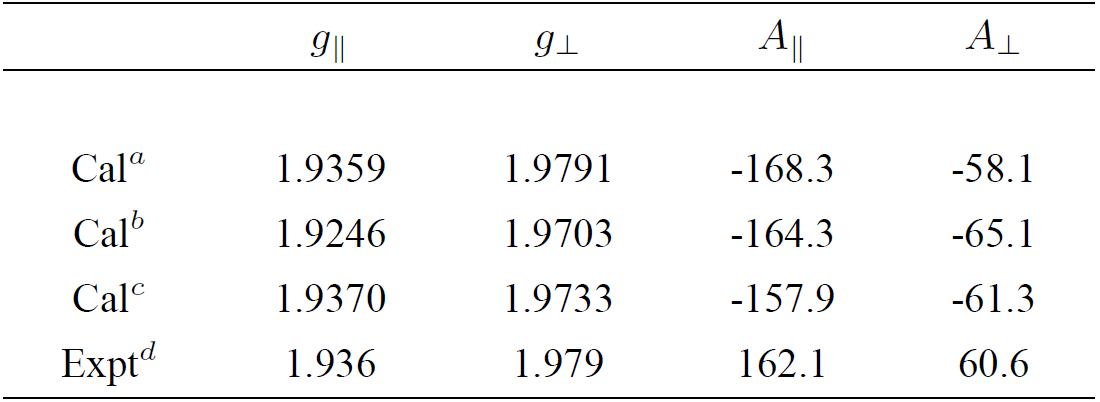
aRef. 7.
bCalculations by using Eqs. (9)-(12) and including the tetragonal distortion in equation (20), but without considering the ligand orbital contributions (i.e., taking λγ=0, ζp=0 and then ζ'=ζ=Nζd, k = k' = N and P = P' = NP
0) in this work.
cCalculations by using Eqs. (9)-(12) and including both the tetragonal distortion in Eq. (20) and the ligand orbital contributions in this work.
dRef. 6.
2. Calculation
As for nd1 ions in tetragonally compressed octahedra, the higher orbital doublet 2eg in original cubic symmetry would split into two orbital singlets 2a1θ and 2b1ε. Meanwhile, the ground orbital triplet 2t2g in cubic symmetry can be divided into an orbital singlet 2
b
2 (ζ) and a doublet 2e(η,ξ), with the former lying lowest 8. Thus, the energy intervals can be written as follows:
E1=E(2e)-E(2b2)=-3Ds+5Dt
(1)
E2=E(2b1)-E(2b2)=10Dq
(2)
E3=E(2a1)-E(2b2)=10Dq-4Ds-5Dt
(3)
Here, D
q
, D
s
and D
t
are the cubic field parameter and the tetragonal field parameters, respectively. They can be calculated from the local geometry of the studied impurity centers and the point-charge model 9,10,11, and be expressed as follows:
Dq=-eq⟨r4⟩/(6R¯5)
(4)
Ds=2eq⟨r2⟩(1/R∥3-1/R⊥3)/7
(5)
Dt=-2eq⟨r4⟩(1/R∥5-1/R⊥5)/21
(6)
Here q(= -2e) is the effective charge of the oxygen ligand. ⟨r2⟩ and ⟨r4⟩ denote the expectation values of the radial wavefunction of d1 orbital in cluster, which can be expressed in terms of the corresponding free-ion values and average covalency factor N with respect to the admixture between the central metal ion and the ligand orbitals 9,10,11:
⟨r2⟩≈3.3222N(a.u.),⟨r4⟩≈21.0698N(a.u.)
(7)
R¯ denotes the average metal-ligand (V-O) bonding length for the studied system (here, we take R¯≈1.985 Å for the VO2+ in cubic case 13). R∥ and R⊥ denote the metal-ligand distances parallel and perpendicular to the tetragonal axis, respectively. For the VO2+ center in CaO-Al2O3-SiO2 system forming tetragonal [VO6]8- cluster, due to Jahn-Teller effect (note: the Jahn-Teller ion V4+ can suffer the Jahn-Teller effect under octahedral environments, which may slightly modify the immediate environment by stretching or contraction of the impurity-ligand bonding lengths), the parallel and perpendicular metal-ligand lengths can be expressed in terms of the average distance and the tetragonal compression ΔR (see Fig. 1) as:
R∥≈R¯-2ΔR, R⊥≈R¯+ΔR
(8)
As mentioned before, previous studies 7 did not consider the contributions to EPR parameters from the s-& p-orbitals and the SO coupling coefficients of the ligands. Thus, using the routines provided in Refs. 14, the high-order (third-order) perturbation formulae of the g tensor and A tensor for 3d1 ion in tetragonally compressed octahedra including above contributions can be obtained based on cluster approach:
g∥=ge-8k'ζ'E2-2k'ζ2E12-4k'ζζ'E1E2-2ζ2E12
(9)
g⊥=ge-2kζE1+kζ2E12-2kζ'2E1E2+2k'ζ'ζE1E2-4ζ'2E22-ζ2E12
(10)
A∥=P-κ-47+P'(g∥-ge)+37(g⊥-ge)
(11)
A⊥=P-κ-27+P'1114(g⊥-ge)
(12)
Where ge≈2.0023 is the spin-only value. The energy denominators
E1 and E2 stand for the
energy divisions between the excited 2b1,
2e and the ground
2b2 states, as mentioned in Eqs. (1-3).
P and P' are the dipolar hyperfine constant
related to the interaction within t2g states and the interaction between t2g and eg states.
The SO coupling parameters ζ, ζ' the orbital reduction factors k, k' and the dipolar hyperfine constant P, P' in Eqs. (9-12) may be written as 15,16:
ζ=Nt(ζd+λt2ζp/2) ζ'=(NtNe)1/2(ζd-λtλeζp/2)
(13)
k=Nt1+λt2 k' =(NtNe)12[1-λtλe+λsA2]
(14)
P=NtP0 P'=(NtNe)1/2P0
(15)
ζd and ζp are the SO coupling coefficients of the free 3d1 and ligand ions, respectively. P0(≈128×10-4 cm-117) is the dipolar hyperfine structure parameter of the free VO2+ ion. A denotes the integral R⟨ns∣∂/∂y∣npy⟩ with the impurity-ligand distance R of the studied system. Nγ and λγ (or λs) are, respectively, the normalization factors and the orbital mixing coefficients for the cubic (O
h
) irreducible representations γ(=eg and t2g) and can be determined from the normalization conditions 15,16
Nt(1-2λtSdpt+λt2)=1
(16)
Ne(1-2λeSdpe-2λsSds+λe2+λs2)=1
(17)
And the approximate relationships 15,16
N2=Nt2[1+λt2Sdpt2-2λtSdpt]
(18)
N2=Ne2[1+λe2Sdpe2+λs2Sds2-2λeSdpe-2λsSds]
(19)
Here Sdpγ (and S
ds
) are the group overlap integrals. N is the average covalency factor, characteristic of the covalency effect (or reduction of the SO coupling coefficient and the dipolar hyperfine structure parameter) for the central ion in the studied system. generally, the mixing coefficients increase with increasing the group overlap integrals, and one can approximately adopt the proportional relationship between the mixing coefficients and the related group overlap integrals, i.e., λe/Sdpe≈λs/Ss within the same irreducible representation eg. Thus, the integrals Sdpt≈0.04553, Sdpe≈0.1198, Sds≈0.0896 and A≈1.0283 can be calculated from the Slater-type SCF functions 18,19 and the metal-ligand bonding length R (≈1.985 Å 13). According to the optical spectra parameter for VO2+ doped in glasses and crystals, 20,21 the average covalency factor N(≈0.81) can be adopted here. Then, the molecular orbital coefficients Nt≈0.8289, Ne≈0.8819, λt≈0.5021, λe≈0.4359 and λs≈0.3271 can be obtained from Eqs. (16-19). And the parameters ζ≈221 cm-1, ζ'≈198 cm-1k≈0.9334 and k'≈0.6893 can be determined from Eqs. (13-15) and the free-ion values ζd≈248 cm-1 for V4+5 and ζp≈151cm-1 for O2-4.
In Eqs. (11) and (12), the core polarization constant k is taken as 0.84, which is the same as κ(≈0.84) adopted in the previous work, 7. Thus, there are only one unknown parameters ΔR in the formulae of the EPR parameters. Substituting the related values (i.e., ζ, ζ', k and k') into Eqs. (9)-(12) and fitting the calculated results to the experimental data, one can obtain:
ΔR≈0.1 Å
(20)
The theoretical results are shown in Table I. For comparison, the calculated results based on the formulae of the EPR parameters within the scheme of the conventional crystal-field theory and various fitted parameters in Refs. 7 are also given in the table.












 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink

