











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink1. Introduction
Theoretical prediction of electronic structure calculations of actinide containing systems remains quite a challenge for quantum chemistry. Several complications prevent standard quantum chemical approaches from being successful in this field in all aspects. On one hand, relativistic and the electron correlation effects become important in heavy elements and should not be neglected in accurate calculations. On the other hand, the presence of several open shells with different main and angular quantum number, i.e. 5f, 6d, 7s, and 7p orbitals are comparable in energies and therefore the bonding can take place with any of these orbitals which add additional complexity for quantum chemical calculations for systems containing these elements.
Many researches have studied the electronic structure of systems containing actinium and lawrencium due to the stability provided by zero and fully populated 5f shell, respectively. In 1998 all electron and valence-only ab initio electronic structure calculations were performed on both actinium and Lawrencium monohydrides and monofluorides by Küchle et al.1. Furthermore, theoretical calculations of actinium monohydride and monofluoride1-5 were carried out at various levels of theory.
Systems with unfilled f electrons, of course, are far more challenging. In fact, actinide oxides have the largest share of study, despite studies of actinide hydrides and fluorides are quite rare. The electronic structure of actinide monoxides were studied by Attila Kovaács and co workers6. In a related work, Ellis et al.7 investigated the electronic structure of actinide monoxides and dioxides by molecular cluster methods based on the first principles one-electron local density theory. On the other hand, a considerable amount of theoretical works8-14 has been devoted to understanding the electronic structure of uranyl ion [U 𝑂 2 ] 2 . In 1983 Krauss and Stevens15 also carried out SCF calculations using a relativistic effective core potential for uranium monohydride and monofluoride and their ions.
Quantum Monte Carlo (QMC) methods are among the most accurate numerical methods to predict relatively accurate properties of quantum systems. Besides its favorable scaling with system size, any arbitrarily complex wave function can be used because the integrals are evaluated numerically. Although there are many different QMC approaches, diffusion Monte Carlo (DMC) remains the most accurate one. Few decades ago diffusion Monte Carlo DMC method has been successful for calculating accurately the ground state properties of many light atoms and molecules. However, QMC applications to actinide systems are very limited. Recently we have tested the performance of the standard B3LYP and the long-range corrected LC-BLYP functionals for both the ground and the excited states of lanthanides and actinides16.Our calculations have indicated that applying the long range corrected scheme to BLYP functional at a value of the range separation parameter 𝜇 equals 0.35 clearly improves the ground state results of 5f actinides.
In this work, and based on our earlier finding, we apply the DMC method employing the LC-BLYP functional at the optimal value of 𝜇 to investigate the electronic structure and bonding of monohydrides and monofluorides of early actinides. Very little theoretical information is available for the monohydrides and monofluorides of actinide elements and the experimental data is still missing. Moreover, to our knowledge this is the first time that a QMC technique has been used to predict the electronic structure of molecules containing 5f-actinides. Only recently, Shi Guo et al.17 have employed quantum Monte Carlo method to study the electronic structure properties for thorium halide molecules.
The basic form of the wave function that we used is the Slater-Jastrow wave function which is considered the most common and simplest one. In the next section, we outline a brief description of the DMC method. The results are then presented and discussed. Finally, we give the conclusion of this work.
2. Computational methods
Diffusion Monte Carlo method has been extensively described in the literatures18-20 so we give here a brief description of it. The diffusion Monte Carlo (DMC) method is a stochastic projector method for solving the imaginary time many-body Schrödinger equation:
where 𝜏 is the imaginary time, 𝜏=𝑖𝑡 and 𝐸 𝑇 is the energy offset.
Importance sampling with a trial wavefunction 𝜓 𝑇 (𝑅) is used to improve the statistical accuracy of the simulation and this is can be achieved by multiplying Eq. (1) by 𝜓 𝑇 (𝑅) and rearranging
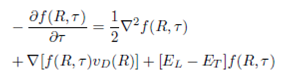 (2)
(2)
where 𝑓(𝑅,𝜏)=𝜓(𝑅,𝜏) 𝜓 𝑇 (𝑅) interpreted as a probability density and 𝐸 𝐿 (𝑅)( 𝐻 𝜓 𝑇 (𝑅)/ 𝜓 𝑇 (𝑅)) is the local energy.
This equation can be simulated with a random walk having diffusion, a draft, and a branching step and may be written in the integral form:
where the Green’s function 𝐺(𝑅,𝑅′;𝛥𝜏) is a solution of the same Eq. (2) and can be interpreted as a probability of transition from a state R to R’. It is possible to use MC method to solve the integral in Eq. (3) but the difficulty is that the precise form of 𝐺(𝑅,𝑅′;𝛥𝜏) is not known. Fortunately the comparison of the Schrödinger equation with the diffusion equation gives us a clue about how one might approximate the unknown Green’s function.
The evolution during the long time interval 𝜏 can be generated repeating a large number of short time steps 𝜏. In the limit 𝜏→0 one can make use of the short time approximation for Green’s function21:
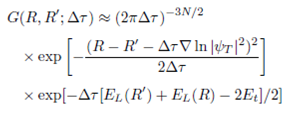 (4)
(4)
But due to the fermionic nature of electrons, the wavefunction must have positive and negative parts and this is opposite with the assumed nature of 𝜓 which is a probability distribution. So the fixed-node approximation22 had been used to deal with the fermionic antisymmetry which constrains the nodal surface of 𝜓 to equal that of the antisymmetric trial wavefunction 𝜓 𝑇 .
In this work, we report predicted calculations of the ground state energies, bond lengths, dissociation energies, and dipole moments for some actinide monohydrides and monofluorides using the DMC method. The Slater determinants were obtained from DFT calculations using the quantum chemistry program Gamess23. We made use of the pseudopotential of Burkatzki and co-workers24 for H and F atoms, which it was constructed for use in QMC and was proved to be quite accurate, while for all actinides CRENBL ECP basis set25 was used. All QMC computations were performed with Qwalk code26. The DMC calculations were performed with a target population of 2000 walkers. These calculations were performed with a time step of 𝜏=0.001 𝐻 −1 which leads to negligible time step error.
3. Results and discussion
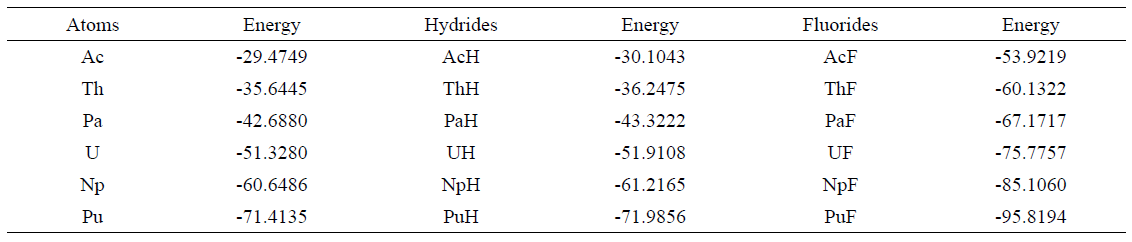
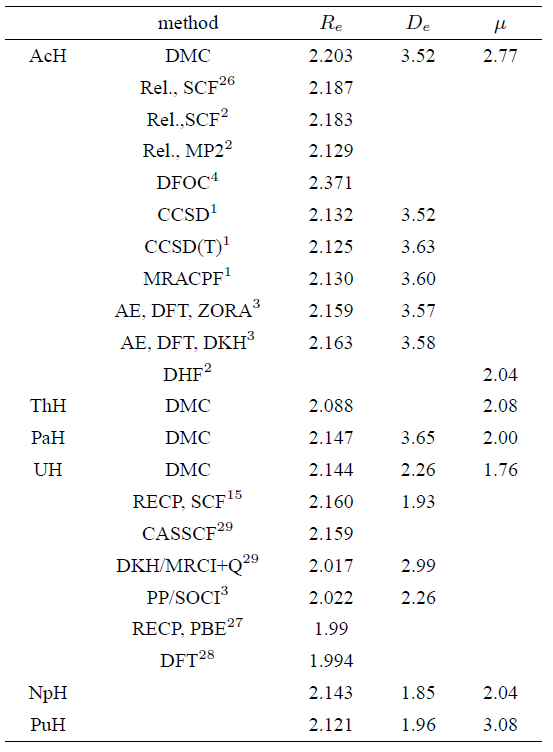
Table I summarizes the DMC ground state energies for actinide monohydrides (AnH) and monofluorides (AnF) computed with the LC-BLYP functional at the optimal value of 𝜇 together with the ground state energies of atoms estimated in our earlier work16. Bond lengths, dissociation energies, and dipole moments for AnH and AnF estimated using DMC along with other computed values available in the literature, are reported in Tables 2 and 3.
Let us first check the monohydrides. From Table II it is obvious that our calculated AcH bond length is in reasonable agreement with that reported by Dolg and also close to Laerdahl et al. value using relativtsting SCF method, but it underestimates the value estimated by Pyykkö via Dirac-Fock One-Centre (DFOC) Calculation4 by about 0.168 Å. On the other hand and based on our result together with the results estimated using DFOC4 and relativistic SCF methods2,26, we suggest that the results cited by Küchle et al. and Hong et al. of AcH bond length seem to be too low. We believe that the long bond length of AcH is most likely due to the involvement of the 6d orbital in bonding with no contribution from the 5f orbital. It is well known that 6d orbital undergoes a large expansion due to relativity which is expected to influence its chemical bonding. In addition, comparing our value of AcH dissociation energy to previous quantum chemical results, our result of 3.52 eV is the same value reported by Küchle and Dolg using CCSD method and is compare well with those found in the other theoretical studies.
Looking at the bond lengths in Table II it can be seen that there are marked shortening of the bond length of ThH which is the shortest bond length across all hydrides under study. In fact, two views were offered for describing the electronic
Table 1 Ground state energies computed within DMC for actinides monohydrides and monofluorides. All energies are in Hartrees.
characteristic of thorium hydride. The first view suggests that Th 5f orbital is particularly filled31-37 and the other view suggests that the Th 6d electrons dominate in bonding38. In light of the significant shortening for either ThH or ThF bond lengths estimated in the present study, we believe that the bonding in thorium is dominating by the 5f orbital which causes this noticeable shortening. Moreover, one can also observe that there is a decrease in bond strength from. AcH to ThH despite the shortening of the bond. In fact, relativistic bond length reduction does not necessarily imply that the bond becomes stronger, as one would expect in the case of bonding in the first row of the periodic table.
From Table II, it is also apparent that there is a small decreasing trend for the bond length of PaH, UH, and NpH molecules, giving evidence of a further transition towards localization of the 5f electrons across the three hydrides. With respect to the dissociation energies, as one might expect, protactinium monohydride has the largest dissociation energy which reflects the fact that Pa atom has the largest electronegativity among all actinides under study. As we will see below, nearly similar effect has been observed for its monofluoride.
Comparing our results of UH with the available reference data, one can see that our estimated U-H bond length (2.144 Å) is much closer to the values provided by RECP, SCF and CASSCF methods than those obtained by the other available theoretical methods. Our estimated dissociation energy of UH is 2.26 eV which is the same value reported using PP/SOCI calculation. Besides, we believe that the dissociation energy of UH obtained with RECP, SCF (1.93 eV) seems to be too low. Unfortunately, there does not appear to be any published work for the monohydrides of Th, Pa, Np, and Pu. It is also interesting to note that the small difference in bond length between PaH and UH, in contrast in the case of fluorides, showing the important role of actinides 6d participation for covalent interactions that becomes more effective at bonding than the 5f orbital. It is worth noting here that several authors [39,40] have suggested the participation of the 6d orbitals in covalent bonding for actinides.
From Table II it is also noticeable that on passing from uranium to neptunium the bond length stays nearly constant, supporting that starting from Np there is a decrease in the 5f participation. This view will be more confirmed and discussed in detail in the case of monofluoride. At the same
Table 2 Bond lengths Re, dissociation energies De, and dipole moments ¹ computed within DMC for actinide monohydrides together with the available reference data.
time we observe unexpected significant shortening of PuH bond length. One might expect a slight contraction for this bond length according to the decreasing trend of the 5f participation across the series. This marked shortening may be probably due to some contributions from the contracted 7s or 7p electrons for PuH bonding particularly the probability of 7s and 7p electrons involvement in chemical bonding increases in the direction Pu⟩Np⟩U. Any way, this effect finds confirmations in some papers. For example, Bing-Yun et al.41 investigated the electronic and structural properties of stoichiometric and non-stoichiometric face-centered cubic Pu hydrides (PuH 𝑥 , 𝑥=2, 2.25, 2.5, 2.75, 3) by employing the Full potential linearized augmented plane wave methods combined with Hubbard parameter U and the spin-orbit effects. They observed a similar abnormal lattice contraction of plutonium hydrides solid which they attributed it to the enhanced chemical bonding and the size effects involving the interstitial atoms. It is also interesting to note that with increasing the atomic number along the actinide series, we notice a considerable drop in D 𝑒 for the monohydrides of Np and Pu which can be interpreted in terms of the strong spin-orbit coupling especially for Pu case which it is well known to weaken the bond.
We now tern to the discussion of the results obtained for monofluorides, which are listed in Table III. Our AcF bond length is somewhat higher than the previous quantum chemical results. This deviation can be explained by the different basis sets used. Again, the long bond length of actinium species can be attributed to significant contribution of 6d orbital which dominants in AcF bonding with no contribution from f orbital. On the other hand, our calculated 𝐷 𝑒 for AcF is 7.07, in good agreement with the all electrons calculations reported by Hong et al.3) using either Zora or DKH calculations however, it underestimates the values reported by Küchle and Dolg.
As can be seen in Table 3, the bond length gets shorter up to UF, and then the trend reverses for NpF and PuF. The shortest An-F distances is obtained for UF which is computed as 2.006 Å lower than the value yielded using density functional theory by 0.016 Å.
Table 3 Bond lengths Re, dissociation energies De, and dipole moments ¹ computed within DMC for actinide monofluorides together with the available reference data.
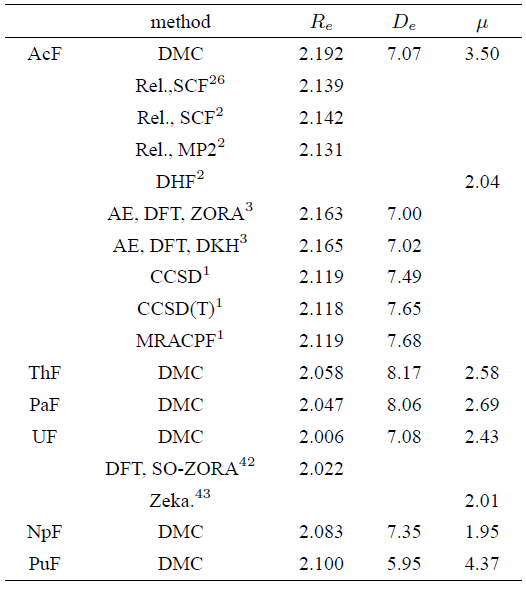
A similar observation has been previously reported by Guellaumont42 using relativistic density functional method on some complexes containing actinides and nitrogen. He found that the shortest bond length is between U and N and he attributed this trend to the presence of covalent effects in the metal-ligand decreases in the orderU ⟩ Pu ⟩ Am ≈ Cm.
Furthermore, Johann Hlina et al. too44 confirmed that uranium-metal bonds are significantly shorter than any other characterized d-f block bimetallic even though ligand flexes to allow a variable U-M separation. The origin of this marked shortening of the bond length up to UF can be traced back to the common actinide contraction which results from a large contribution of 5f-electrons to bonding. One can conclude from these results that the U 5f orbitals play the primary role in the ionic bonding in line also with the DFT calculations of Minasian et al.45 who predicted a large 5f contribution to bonding in UCl 6 −2 and UOCl 5 −1 . At this point it is important to point out that our result for UF provides additional confidence to the significant U 5f orbital participation in bonding especially in the case of fluorides due to the strong 5f overlap with the high electronegative fluorine.
Another important effect to note in Table 3 is the unexpected increase in the NpF and PuF bond lengths. The reason of this effect originates entirely from the decreasing contribution of 5f electrons to bonding. From our point of view, even in the case of small participation of 5f-electrons in bonding, the actinide contraction is negligible since the An-F bond is more rigid than An-H bond which causes the observed increasing in the bond length particularly in the case of PuF. A previously theatrical study of Moritz et al.46 supports our view who explained the minor actinide contraction in the actinide-fluorine bond length than their counterpart in actinide-oxide to the more rigidity of An-F bond. A similar behavior has been also observed by Wang and Schwarz in their paper47 on some lanthanide diatomics. Their density functional calculations for lanthanide monohydrides, monofluorides, and monoxides found that compounds with rigid bonds undergo only a small contraction.
From our calculations for the An-F it does appear that Np has a fractional emergence of localization in the 5f states, even though the widespread assumption that Th to Pu have delocalized 5f states and Am to Lr have localized 5f states. In fact, there are some researchers have given results for Np and Pu that are in agreement with ours. In 1984, Brooks et al.48 showed that while the 5f spin-orbit interactions can be neglected in the lightest actinides, they become important for Np. In other words, very beginning of the transition from LS coupling to intermediate coupling occurs in Np, which means a fractional appearance of localization in Np.
A further confidence to our observation has been provided by Moore and Laan49. They proved that the localization begin to appear in 5f states in Np by using the values of actinides bulk moduli. The experimental bulk modulus of each of the light actinides is: Th at 50-72 GPa50-52, Pa at 100-157 GPa51,53,54, U at 100-152 GPa55,56, Np at 74-118 GPa51,52,57, Pu at 40-55 GPa51,52,58, Am at 30 GPa59. Notably, Th exhibits low bulk modulus due to the small amount of electrons in the 5f states whereas, Pa and U exhibit the highest bulk moduli but there is a drop appears in Np which they related it to a fractional emergence of localization in the 5f states.
However, as we have noticed above, for the monohydrides there is a gradually decreasing in the bond length across the series with very small difference between UH and NpH bond lengths. This is an expected consequence of the difference in electronegativity between hydrogen and fluorine atoms. The strong inductive effect of the electronegative fluorine strengthens the bonding in the monofluorides and becomes able to get rid of the small contraction in NpF and PuF molecules resulting from the common actinides contraction.
Now it remains to clarify our estimated values for the dipole moment. Note that going from the monohydrides to the monofluorides the bonds become more polar which of course increase the dipole moments of monofluorides than monohydrides. It is clearly seen from Tables 2 and 3 that this consistents with all estimated dipole moments with exception of Neptunium. Our calculated value for NpF is smaller than NpH by about 0.09 D.
It is also worth noting here that for both the monohydrides and monofluorides we find that the dipole moments of actinium and plutonium species are greater than those of other actinides. As we know the dipole moment monotonically increases with the bond lengths corresponding to the growth of the distance between the effective charges. Consequently, the somewhat large dipole of species containing actinium is not a surprise which is traced back to the long bond length observed in both the monohydride and monofluoride. Moreover, the dipole moments of plutonium species being the highest among all molecules under study which can be interpreted in terms of its different electronic configuration. Atomic Pu has a 5f 6 7s 2 configuration in its ground state which differs from the earlier elements having partial occupation of the 6d shell besides 5f shell. Given the fact that f-electrons are less polarizable than d-electrons, one can expect this increasing trend in dipole moments for all Pu species.
Finally, it should be remembered that all calculations reported here were performed using only Slater-Jastrow functional. Despite the simple nature of this type of functional, our results are able to give a reasonable description of the electronic structure of early actinide monohydrides and monofluorides. In light of these considerations, we can safely confirm that the DMC method employing LC-BLYP functional at the optimal value of 𝜇 is a good choice for future study of actinide containing systems
4. Conclusion
We have calculated ground state total energies, bond lengths, bond dissociation energies, and dipole moments for early actinide monohydrides and monofluorides using the diffusion Monte Carlo (DMC) method and LC-BLYP functional. The present study shows that U-F bond length exhibits the shortest actinide-fluoride distance, supporting results from previous investigations. However, the bond length of the ThH is the shortest among all hydrides under study. Our calculations also provide strong evidence that neptunium is the first actinide element having a small degree of localization in the 5f states in line with some previous studies. Moreover, dipole moments of plutonium species being the highest among all molecules under study which consistent with its different electronic configuration with no 6d orbital occupied. On the whole, the present work demonstrates that the DMC method employing LC-BLYP functional at the optimal value of the range separation parameter can be extended to calculate the electronic structure properties of actinide containing systems.

