











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introduction
The knowledge of physics parameters of semiconductor compounds is of great interest in order to evaluate their potential use in a wide class of optoelectronics devices. These parameters have been investigated not only experimentally, but also theoretically. From the standpoint of the latter, crystal lattice energies are important in considering the stability of new materials and reflect the natural tendency towards the organization of matter1; meanwhile the energy gap is important to determine the region of the light spectrum where the semiconductor can be applied like photonic device.
There is a large practical motivation for the study of magnetic semiconductors due to their potential for combining semiconducting and magnetic behavior in a single material system. Magnetic semiconductors are viewed as enabling materials for magneto-electronic devices and technology.
In 1955 Hahn et al. synthesized a new family of compounds of type II-III 2 -VI 4 from the mixture of (II-VI) + (III 2 VI 3 ). Compounds of this family are characterized by a variety of crystalline structures according to the composition cation-anion and to the position of the cations in sites with tetrahedral or octahedral coordination.
The ternary compounds of the family II-III 2 -VI 4 , depending on the number of tetrahedral and octahedral sites for cations, may crystallize in three main structures: cubic spinel type, rhombohedral and tetragonal defective. The term defective refers to certain atoms of the unit cell that have not orbital linked and directed towards a point in the lattice called vacancy2,3.
Most ternary semiconductor of the family II-III 2 -VI 4 (II are the divalent cations Mn, Zn, Cd and Hg; III are the trivalent cations Ga, In and Al, and VI are the chalcogens S, Se and Te) crystallize in the tetragonal structure of chalcopyrite defective or stannite type4-6. These compounds, except those which have Hg as divalent cation, have been widely studied because they exhibit interesting optical properties such as high photosensitivity, intense photoluminescence and high optical activity; all this properties combined with a large energy gap (up to 4 eV) make them potentially attractive source materials as photodetectors of visible light and ultraviolet7.
The MnGa 2 Se 4 has both semiconducting and magnetic properties which make it a good candidate for applications such as optoelectronic devices. The properties of this compound has been studied since the 80s due to their use in storage devices as well as devices that require magneto-transport and magneto-optical8 properties.
In this paper we present a theoretical study by using the Crystal 06 software and ab initio Density Functional Theory (DFT) and Hartree-Fock (HF) methods, with different base functions, to calculate the total energy and the energy gap of the magnetic semiconductor compound MnGa 2 Se 4 .
2. Methodology
All the calculations of crystal structure, lattice energy and energy gap of the MnGa 2 Se 4 compound have been performed using the software package Crystal 06 [9] which is an ab initio program based on linear combination of atomic orbitals for the treatment of periodic systems. In each case the fundamental approximation made is the expansion of the single particle wave functions, named Crystalline Orbital, as a linear combination of Bloch functions defined in terms of local functions, indicated as Atomic Orbitals. The local functions are, in turn, linear combinations of Gaussian type functions whose exponents and coefficients are defined by input9.
The software has two visors or interfaces, one of which was developed by the same designers of the software package and requires supplement it with some external executables and DLV interface10, available in complementary to Crystal, that, although it is not located in the program, lets us view output files.
The calculations were performed assuming room temperature (273 K), no external forces acting on the compound and keeping the number of cycles and convergence parameters for the UHF (Unrestrictre Hartee-Fock) antiferromagnetic state case.
The atomic positions, used as initial conditions for the DFT and UHF MnGa 2 Se 4 analysis were taken from Ref. 3.
The steps to perform the calculations are as follows:
The molecular structure, space group and crystallographic parameters of the compound MnGa 2 Se 4 are specified. Moreover, the spatial coordinates of the constituent atoms and their atomic numbers, as well as the vacancies are established, generating a superlattice.
We selected a set of base functions allowed by the software package Crystal 06 to calculate the energy of the compound MnGa 2 Se 4 at enviroment conditions.
The Hamiltonian and convergence functions are selected.
END keyword closes each of the phases described.
If the phases are written correctly, and the bases converge, the system starts the running of the test; otherwise the system reports an error, in which case we have to adjust the parameters of the base again or select alternatives bases.
The system allows the generation of superlattices and other geometric characterization parameters. In our case, we used a double superlattice configuration for antiferromagnetic compound.
Table 1 shows the parameters used for the first block.
The lattice parameters were taken from Ref. 8 and the atomic positions from Ref. 3. The second block contains the used base, followed by the commands BASE, 99 0, END. The third block contains the convergence functions and the used Hamiltonian: HF or DFT UHH.
The choice of the set base is a fundamental step in defining the level of calculation and accuracy. This is of particular importance in the study of periodic systems where there is a large variety of chemical bonds.
One of the main objectives of the ab initio calculations is the accuracy and this procedure is a balance between accuracy and costs. Consequently, we should use good set bases
in pursuit of quality despite its computational cost to avoid producing results without physical meaning.
The selected bases, prior analysis of their functionality or running under Crystal 06 are: Pople STO-3G12, Pople 3-21G12, Ahlrichs VTZ12 and Stuttgart-Dresden pseudopotentials13.
3. Discussion and analysis of results
The MnGa 2 Se 4 is an antiferromagnetic compound5 which crystallizes in a defective chalcopyrite tetragonal structure with space group 𝐼 4 and lattice parameters 𝑎=5,671 Å; 𝑐=10.754 Å; 𝑐/𝑎=1.896 Å 13, 𝑎=5.686 Å; 𝑐=10.760 Å;8 𝑐/𝑎=1.892, 𝑎=5.665 Å; 𝑐=10.754 Å; 𝑐/𝑎=1.895 Å 14.
Figure 1 shows the tetragonal unit cell of MnGa 2 Se 4 , at room temperature, generated with the parameters specified in Table I and visualized with the DLV interface. Table 2 shows the values of the lattice energy calculated using different bases for the MnGa 2 Se 4 compound.
Table 2 Lattice total energy of the MnGa2Se4 compound calculated by using the UHF-antiferromagnetic method.

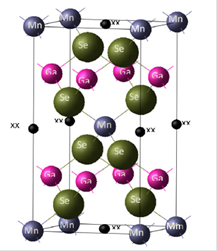
Figure 1 Tetragonal unit cell of the MnGa2Se4 compound at room temperature (space group ¹I4). XX refers to a vacancy.
By using the Stuttgart pseudopotentials the obtained value for the lattice energy of MnGa 2 Se 4 is up to two orders of magnitude different from that obtained with other base functions. For this compound, we did not find any published experimental data about the lattice energy with which we can compare the obtained values and verify the effectiveness of this method of analysis. However, it seems that Stuttgart pseudopotentials produce the best result. The obtained value with this base function, 5.84 eV (563.47 KJmol −1 ), is comparable but less than the lattice energy of ionic compounds, which also are characterized by high melting points. The larger the lattice energy, the more stable the solid and the lattice energy is reflected in the melting point of the compound. In the case of the MnGa 2 Se 4 no melting point was found in the literature, but for others II-III 2 -VI 4 defective chalcopyrite tetragonal compounds, with the same space group of MnGa 2 Se 4 , have been reported melting points from about 700 ∘ C to 1400 ∘ C, approximately15.
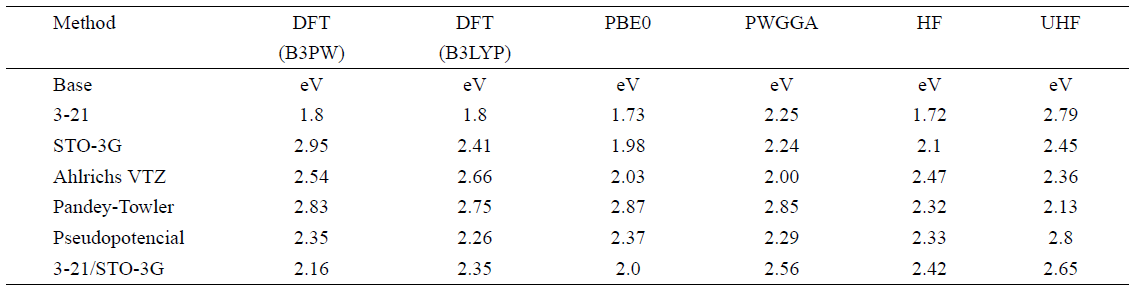
Table 3 Base Functions and gap energies calculated for the compound MnGa2Se4 using HF and DFT methods with the Crystal 06 program. The base 3-21/STO-3G is the combined basis STO-3G + 3-21.
3.1 Energy gap of the MnGa 𝟐 Se 𝟒 compound
Many authors have published studies on the band structure and on the value of the energy gap of the compound MnGa 2 Se 4 . It was established that the compound is a direct energy gap material with reported values of 2.31 eV6,7,16, 2.33 eV17 and 2.77 eV18. All these values indicate that there are still significant differences in the value of the energy gap.
Table III shows the results obtained by applying the ab-initio Hartree-Fock methods for the energy gap of MnGa 2 Se 4 by using the base functions 3-21, STO-3G12, Ahlrichs VTZ11, Pandey-Towler9, pseudopotential and 3-21/STO-3G11. Some of these base functions are available on the program page and others were obtained by converting base 94 Gauss11, using the Gausstcrip9 software, available on the website of Crystal 06:
The values of the energy gap reported in the literature for MnGa 2 Se 4 vary from 2.31 eV5,6 up to 2.77 eV17. As shown in Table 3, except for the base 3-21, all bases reproduced energy gap values which are between the experimentally measured values. However, again, the range of values reported in the literature does not enable a comparison that describes the effectiveness of the applied method.
It was showed19 that the optical transitions in II-III 2 -VI 4 compounds, like ZnGa 2 Se 4 , occur at the center of the Brillouin zone. Since MnGa 2 Se 4 is an isoelectronic analog of ZnGa 2 Se 4 , it can state20 that in MnGa 2 Se 4 the narrowest energy gap is located at the center of the Brillouin zone 𝛤(000). In the region of the fundamental absorption edge, the direct optical transitions, at 295 K, occurs at 2.5 eV20.
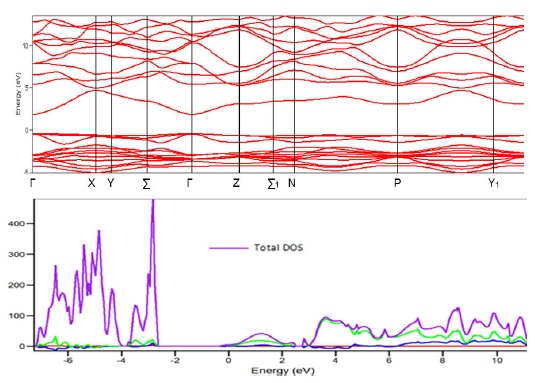
Figure 2 shows the band structure of MnGa 2 Se 4 compound along the lines of high symmetry in the Brillouin zone and the density of states (DOS) calculated by using the DFT method and B3PW base pseudopotential function. As can be seen, the maximum of the valence band and the minimum of the conduction band are located at 𝛤-point so the MnGa 2 Se 4 is a direct gap compound, as stated by Tagiev et al. The energy gap value calculated using this method is 2.35 eV.
4. Conclusions
Applying the Hartree-Fock and DFT computational methods with the Crystal 06 package is possible to calculate the energy of the lattice structure and the band gap energy of the magnetic semiconductor MnGa 2 Se 4 compound, using different basis functions. However, due to the lack of knowledge about the lattice energy and to the several values of the energy gap reported in the literature for the compound under study, it is not possible to determine which bases faithfully reproduces the experimental values of MnGa 2 Se 4 . Nevertheless, by using the DFT method and B3PW base pseudopotential function, it is possible to calculate the band structure of the compound which is consistent whit the experimental results.

