











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkPACS: 32.80.Rm; 32.80.Wr; 33.15.Ta
1.Introduction
Different sources contribute to the formation of acetaldehyde: anthropogenic, plants, biomass burning, surface of the oceans, being its largest source, the oxidation of hydrocarbons. Photochemistry of acetaldehyde had been previously analyzed; it can be decomposed by photolysis 1. The interaction of acetaldehyde with the radical HO allows the hydrogen abstraction to produce the acetyl radical, and with ions and radicals such as HO and NO2 produces peroxyacetyl nitrate (CH3C(O)O2NO2). Acetaldehyde has been detected during combustion processes as a part of radical reactions 2, is sensible to oxidation processes, resulting in the formation of acetic acid 3. It also has been studied using different experimental and theoretical methods4,5,6,7,8,9. The electronic, rotational and vibrational structure has been reported10,11,12,13,14. Electronic energy levels and associated vibrational progressions have been measured using various techniques 15,16. Due to the fact that acetaldehyde is a small molecule, the analysis of experimental data can be accomplished comparing them with quantum chemical calculations15,16,17,18. Previous results show that when the acetaldehyde molecule absorbs photons in the UV wavelength range, between 250 and 350 nm 19 the transition S0 → S1 occurs through a
The energies of the first excited electronic states of acetaldehyde are shown in Table I.
Table 1 Excitation energies of acetaldehyde.
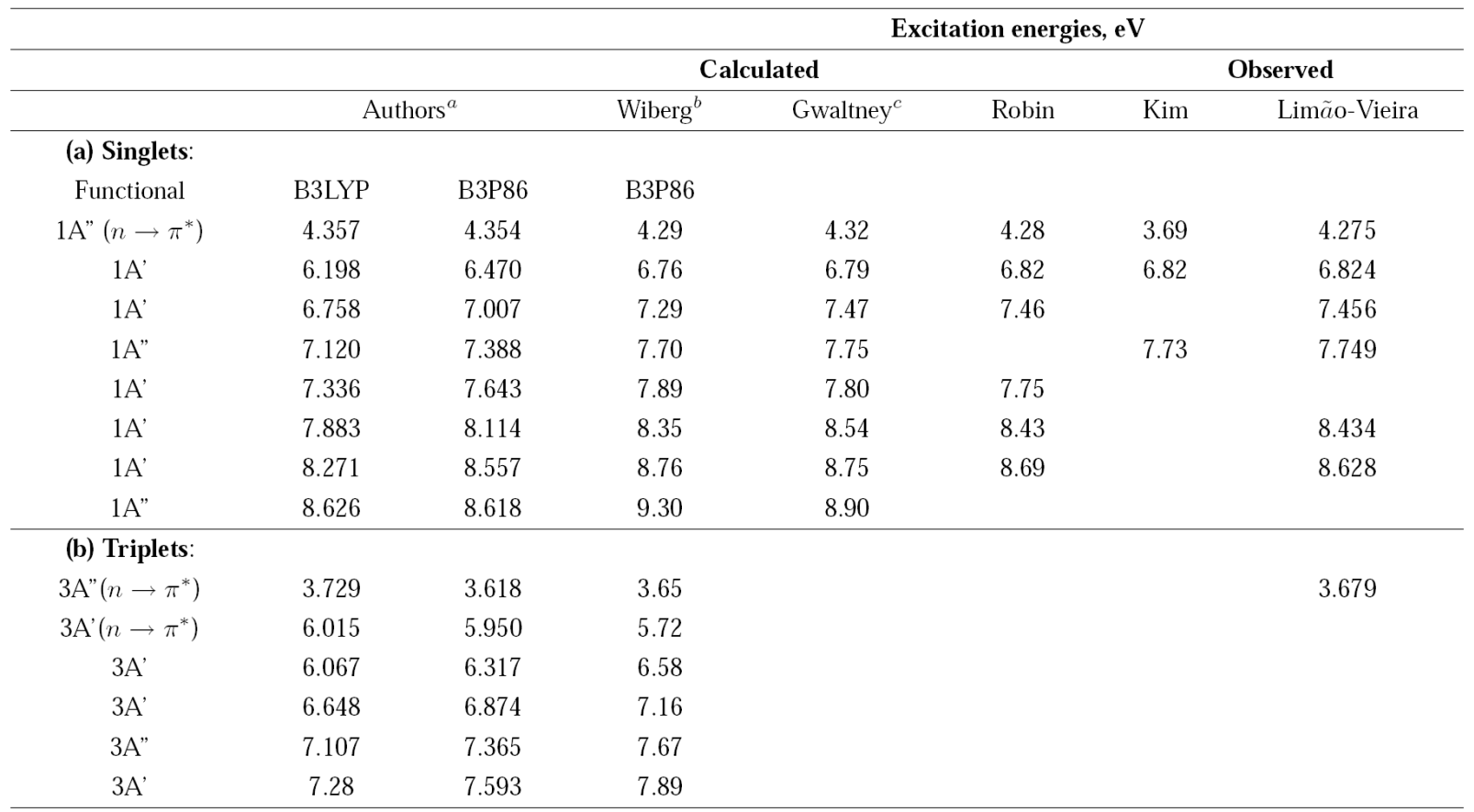
a Time-Dependent Density Functional Theory, Functionasl B3LYP and B3P86, 6-311++G(3df,3pd) basis set.
b Time-Dependent Density Functional Theory, Functional B3P86, 6-311++G(d; p) basis set.
c Equation-of-Motion Coupled Clusters Method, [5s3d2d/3s2p] basis set.
The accessible dissociation channels by absorption of a single photon at wavelengths below 318 nm (3.89 eV) are:
The channel (2) is possible through the T1 state by an intersystem crossing process (ISC) with the S1state 24,1. The radicals CH3 and HCO can result from the dissociation of the acetyl radical after hydrogen abstraction 25,26. In the S1 state, the acetaldehyde can dissociate to form hydrogen and the CH3CO radical in an excited state, following the channel (3). Furthermore, as it has been reported 27, CH4 and CO must arise from the non-degenerate singlet ground state S0, channel (4),
Other important channel is the hydrogen elimination channel (5):
The channels (3) and (5) are also a result of an ISC mechanism, between the S1 and T128.
The molecular products CH4 and CO are obtained through channel (4) at wavelengths shorter than 248 nm, while the hydrogen atom elimination channels (3) and (5) emerge from the photodissociation of acetaldehyde at 205 nm 17. Channel (3) dominates over channel (5) due to the different transition energy values: 24.4 and 42.5 kcal-mol. Other reported channel 29, which leads to hydrogen neutral molecule elimination, is:
All of the above processes, channels (2) to (6) can take place in one photon absorption regimes and the resulting neutral products have been reported. For instance the absorption of one-photon of 266 nm allows the neutral molecule reach the S1 state. If there is not an excess of energy transferred during the excitation, the S1 state can decay to S0 by an internal conversion processes IC, and form other products 29,30, including CH4 and CO.
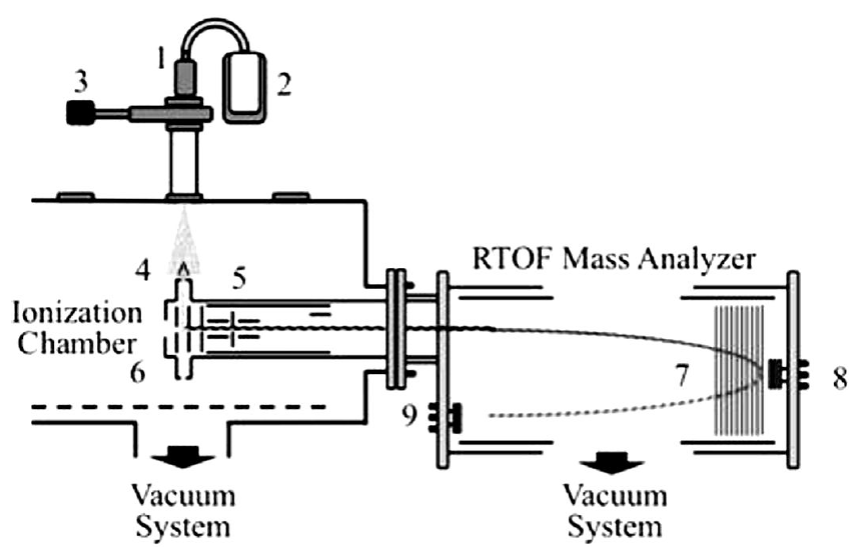
In the present work, the photodissociation and photoionization of acetaldehyde in the multiple photon absorption regimes were investigated at wavelengths of 266 and 355 nm and intensities of radiation in the range 108 to 1011 W⋅cm-2. Radiation interacts with a cooled molecular beam of acetaldehyde produced by the adiabatic expansion of vapors into a high vacuum chamber at 10-8 torr. The resulting ions were analyzed using a home-assembled Jordan R-TOF mass analyzer.
On the basis of the detected ions at 266 and 355 nm, the processes were identified as D-I and I-D, respectively. The number of photons required to form a particular ion was calculated accordingly with the Keldysh approximation 31 and compared with the reported electronic energy levels 32,8. Along with those, based on the detected ions, the different dissociation pathways were proposed.
This work gives new insights on the molecular physical processes present when acetaldehyde molecules interact with photons and the products that can emerge as result of photoionization and photodissociation.
2. Experimental
The photoionization and photodissociation of acetaldehyde was analyzed using the experimental setup previously described 33. Briefly, a sample of acetaldehyde ((99%), purchased from Sigma Aldrich Chemical Corp) was used as received. The sample was connected to a pulsed injection valve, IOTA-ONE. A cooled molecular beam of acetaldehyde, with helium as a carrier gas was produced by adiabatic expansion in a high-vacuum chamber at 2 x 10-8torr. The pulsed valve was synchronously coupled with the laser pulses with an opening time of 250 μs to achieve an operating pressure of 2 x 10-6 torr. The 355 nm laser radiation was produced from the third harmonic of a Nd:YAG laser, operating at 30 Hz repetition rate (Spectra Physics). The laser pulse width is 5.5 ns and the energies per pulse from 1 to 30 mJ. When it was required, 266 nm photons were produced from the fourth harmonic by pumping a second crystal, frequency doubler, with 532 nm pulses of radiation. Thus, 266 nm photons with pulse widths of 3.5 nm and energies per pulse from 0.1 to 10 mJ were used. The laser radiation (with a Gaussian profile and linearly polarized) was focused into the interaction region using a 15 cm focal length lens. The diameter at the focal point was 80.0 μm. Under these experimental conditions, radiation intensities between 109 and 1011W⋅cm-2 were achieved. The cooled molecular beam interacted orthogonally with the laser radiation at a point located between two parallel plates continuously polarized at 5.0 and 4.5 keV, corresponding to the extraction and the acceleration potentials, respectively. The distance between the plates was 0.8 cm. Holes of 10 mm diameter at the center of each plate with a fine metal mesh were used to extract the positively charged ions from the interaction region. The ions were driven along the field-free region of the reflector R-TOF analyzer and eventually reached a double microchannel detector after they were refocused; Figure 1 shows the experimental setup. The ions arrived to the detector, according to their masses. The resolution archived was of the order of 3000. The current signal was pre-amplified, voltage-converted, digitized and sampled in time using an EG&G Ortec multichannel analyzer. The resulting detector signal after each laser pulse was processed using a temporal window of 50 μs and 80000 channels at 0.625 ns per channel. Signals from 5000 laser shots were added to obtain the final spectra. Spectra were obtained at different energies per pulse in the intervals from 0.1 to 10.0 mJ at 266 nm and 1.0 to 20.0 mJ at 355 nm.
3. Results and Discussion
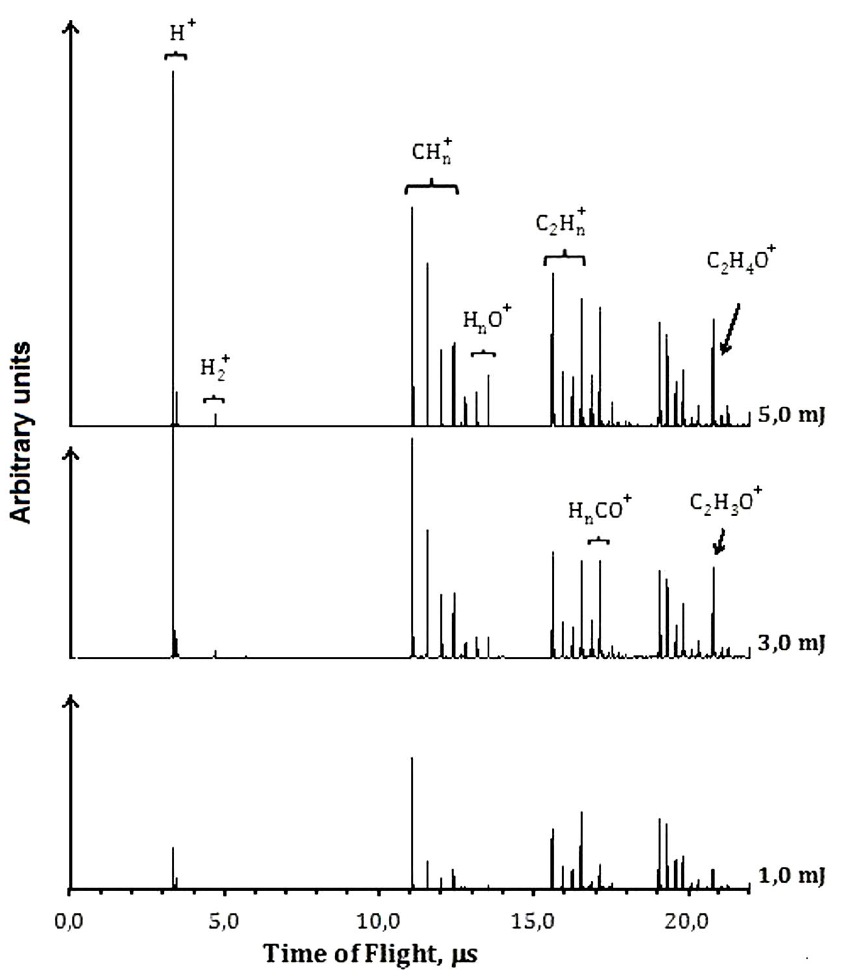
The ions from PI and PD process were characterized by R-TOF mass spectrometry technique. The spectra were measured at different energies per pulse. Each ion current was analyzed as it changes as a function of the energy per pulse. When the photon density increases, new ions can be formed, and the TOF mass spectra changes, see Figs. 2 and 3. If two photons of 266 nm (9.32 eV) are absorbed, it results in the dissociation of the molecule along the C(O)-H coordinate to form the CH3CO, represented as channel (3), and the ions are formed later. The photophysical processes are mainly dissociation-ionization since only very low current of the parent ion was detected. The acetyl radical absorbs photons and gets ionized and dissociated Fig. 2. The main ions detected in the TOF mass spectra correspond to CH3CO+ (43), HCO+ (29), CO+ (28),
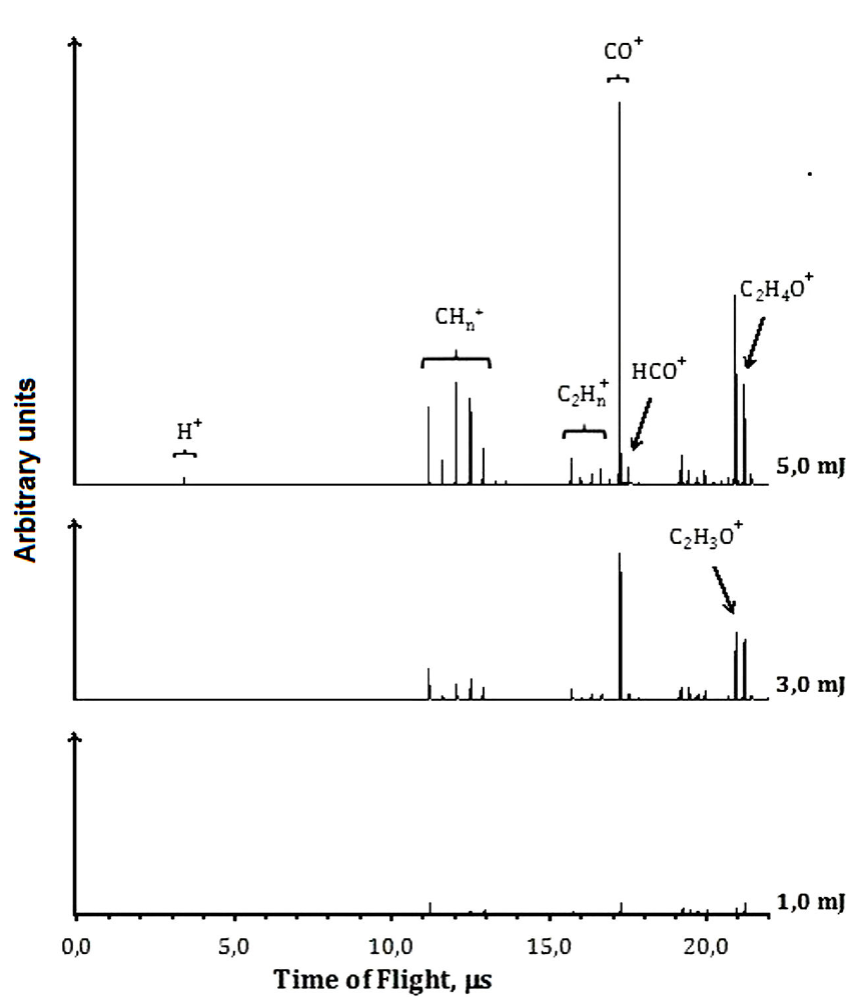
At 355 nm and low energies per pulse, such as 1.0 mJ, the molecular ionization or dissociation processes are poorly observed. Increasing the energies, as to 3.0 mJ per pulse, the presence of the molecular parent ion,
The presence of
Hydrogen cleavage to produce H+ is a non-favored channel; H+ is detected only in very low quantities. Contrary to the case at 266 nm, the ions resulting from vinyl alcohol dissociation were not present.
3.1. Energies of the Processes
For multiple-photon absorption, the number of photons and from them the energy for the formation of a particular ion via ionization or dissociation processes can be calculated using the ion currents measured as a function of the radiation intensity, applying the Keldysh relation, Y=A⋅ I n 31. Where, Y is the ion yield of a particular ion, I is the intensity of radiation in W⋅cm-2, n is the number of photons for the formation of the ion, and A is a function of the nth order cross section. At low energies per pulse when the process follows the Keldysh’s relation, the value n can be readily calculated. As the radiation intensity increases, it is possible to reach the saturation limit, and the calculated values of n decrease. Calculated n values and the equivalent energy for the main detected ions of acetaldehyde at 266 nm and 355 nm are shown in Table II along with their ionization energies, previously reported 34.
Table II Calculated energy values, in eV, from the number of photons absorbed using the Keldysh approach 31.
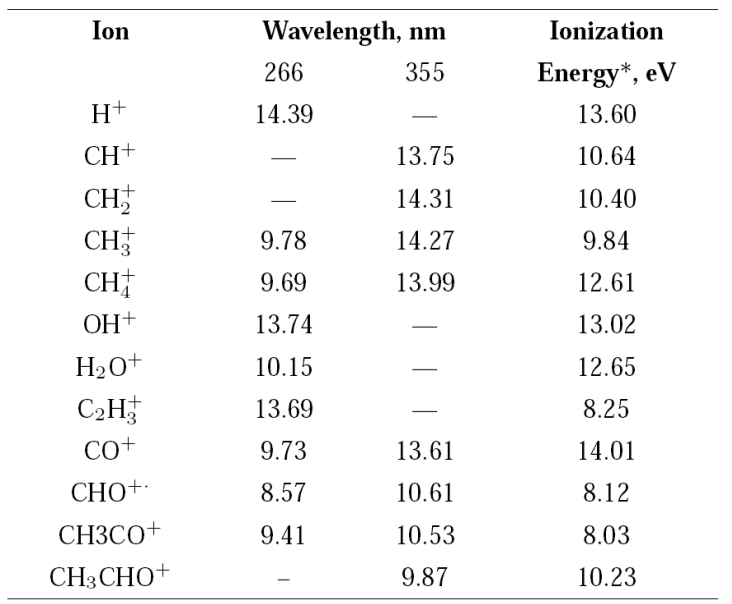
*From reference 37.
Most of the energies calculated from the number of photons leading to a particular ion are in good agreement with the reported values. In some cases, the number of photons n gives the energy to form a particular ion coming from the neutral acetaldehyde or from the acetyl radical produced after the acetaldehyde dissociation, Table II.
3.2. Dissociation-Ionization at 266 nm
Accordingly to Table I the absorption of two 266 nm photons allows the neutral acetaldehyde molecule access different electronic states. The energy of one photon exceeds the S1 state, but it is not enough to reach the S2 state, however, at high photon densities such as in the present experiments, two-photon absorption is possible. The two photon supply an energy of 9.32 eV and electronic transitions to molecular Rydberg states occur. The excited molecule can decay to the triplet state T2, along the channels
The formation of
The formation of the radical HCO +⋅ at 266 nm, only takes place by one mechanism, the molecular dissociation channel (2), followed by ionization. The energy calculated from the number of absorbed photons leading to the ionization of HCO ⋅ at 266 nm is in good agreement with the reported ionization potential of 8.12 eV 37, see Table II.
CO+ arises from two different mechanisms, by dissociation of acetyl cation, channel (9), and from dissociation of HCO radical cation, follow by ionization, channel (10).
Finally, the formation of H+ results also from two different processes, single dissociation: C-H, bond breaking, from cations such as CH3CO+, channel (11), and HCO+, channels (12), Fig. 2.
For the main dissociation channels, the C-C bond dissociation that produces CH3 and HCO radicals requires an energy near 4.15 eV, while the C-H bond dissociation that produces CH3 CO and H, requires 3.85 eV 28.
Figure 4 shows the ratio of ion currents
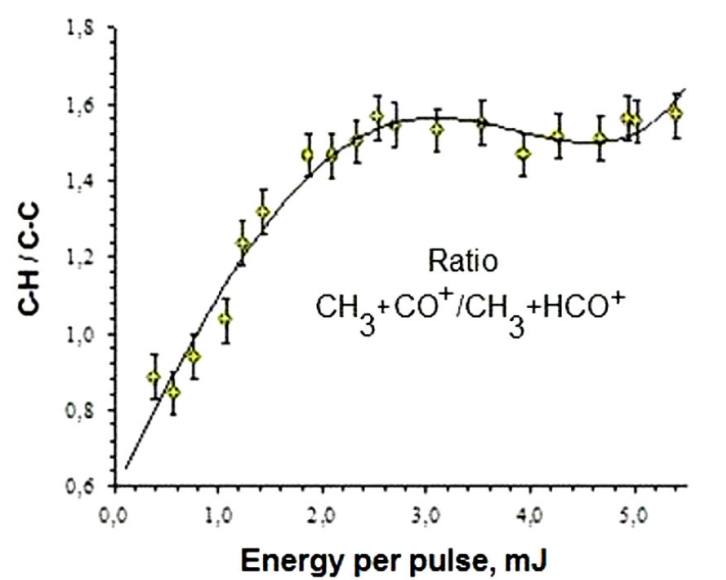
3.3. Ionization-Dissociation at 355 nm
At 355 nm, based on the detected ions, we can propose a mechanism for the formation of ions identified in the TOF spectra, see Fig. 3. The absorption of one 355 nm photon (3.493 eV) does not supply enough energy to reach the first excited state S1 (4.29 eV,
Three photon channels:
Hydrogen elimination:
C-C bond dissociation:
Four photon channels:
Hydrogen elimination:
C-C bond dissociation:
Hydrogen transposition via the roaming mechanism:
Double bond dissociation:
We suggest that the formation of the CO+ cation could be produced through channel (19) or by the dissociation of
If
From a theoretical point of view, the C-H channel dominates over the C-C channel when the molecular parent ion dissociates. Experimentally, the ion yields of
3.3. Intramolecular hydrogen transposition
At 266 nm, the spectra were characterized by the appearance of fragment ions different from those obtained by the direct DI process of acetaldehyde. Such ions are the result of the dissociation of the ionized vinyl alcohol. The ratio of the ion currents resulting from the dissociation of vinyl alcohol and acetaldehyde (
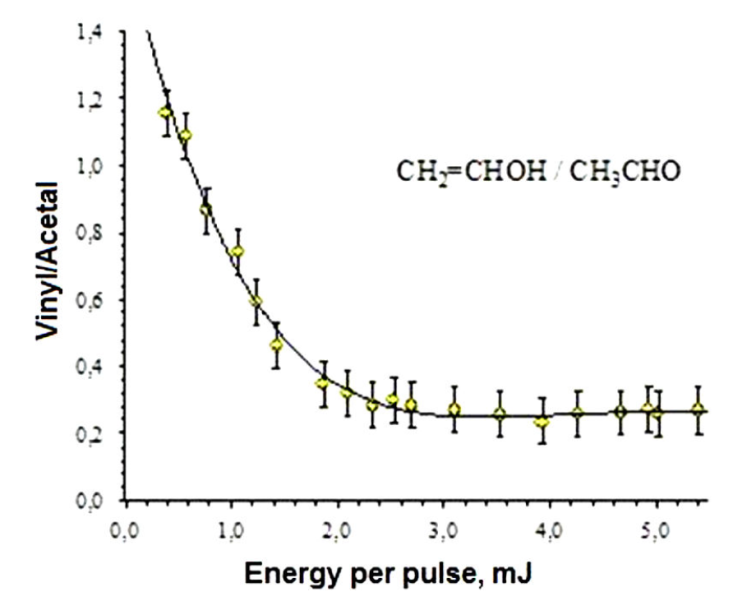
As it was previously mentioned, the energy barriers for enolization were calculated for the neutral and cation forms of acetaldehyde. In the neutral form, the energy barrier (3.00 eV) is higher than that calculated in the cation (2.71 eV) by approximately 0.29 eV 21,12. In the cation, the equilibrium is displaced to the enol (vinyl alcohol) because the alcohol has a lower energy than acetaldehyde, with an energy difference of about 0.48 eV. These results are in agreement with of ion yields at lower energies per pulse, see Fig. 5.
At 355 nm there is a resonant process, the neutral molecule is ionized, however only their daughter ions were observed in the TOF spectra, see Fig. 3. Excess of energy allows the equilibrium to the formation of vinyl alcohol cation, their daughter ions were observed, but not as efficiently as it occurs at 266 nm.
4. Conclusions
Multiphoton I-D D-I studies are an excellent tool to address the main process that take place when photons interact with neutral species. In the present work, it is reported the analysis of photoionization and photodissociation of acetaldehyde using laser radiation of 266 nm and 355 nm in a cooled molecular beam. TOF-MS spectra are presented. At 266 nm, photon absorption is followed by a D-I mechanism, while at 355 nm, the I-D mechanism occurs. Some new dissociation channels were proposed based on cations detected from the I-D or DI processes.
At 266 nm, the spectra show a signal attributed to the dissociation-ionization of vinyl alcohol, which arises from the gas phase molecular equilibrium of the acetaldehyde transformed into to vinyl alcohol. For the case of 355 nm a cuasi-resonant processes is observed and there are several differences with the 266 nm spectra.

