











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCTION
Neutropenia is a wide-range hematopoiesis disorder characterized by granulopoiesis arrest at the promyelocyte maturation stage with peripheral blood absolute neutrophil count below 1500/µL or below 500/µL in severe neutropenia, for a normal range of 2500-6000/µL. Severe congenital neutropenia (SCN) includes a heterogeneous group of disorders of hematopoiesis deficient mature neutrophils1. SCN type 4 (SCN4) is a rare disease characterized by the presence of SCN, associated with non-hematologic multisystemic traits, that was simultaneously described by Dursun et al. (2009) and Boztug et al. (2009), identifying both groups of authors the underlying genetic cause2,3. The characteristic non-hematological manifestations include increased visibility of superficial veins (IVSV), congenital heart defects, and urogenital anomalies. In addition, less frequent findings are thrombocytopenia, failure to thrive, inner ear hearing loss, and endocrine disorders such as growth hormone deficiency, delayed puberty, and cutis laxa4,5. As a group, SCN has an estimated prevalence of 6/1,000,000 individuals; in contrast, SCN4 has a prevalence of 1/10,000,000 people6. The gene G6PC3, located in 17q21, encodes the ubiquitously expressed glucose 6-phosphatase-β enzyme (G6Pase-β or G6PC3)7,8.
G6PC3 catalyzes the final step of glycogenolysis, hydrolyzing the glucose-6-phosphate to glucose and inorganic phosphate in the endoplasmic reticulum (ER) (4). The biallelic G6PC3 gene variants trigger energy homeostasis impairment, failure in 1,5-anhydroglucitol-6-phosphate elimination, ER stress, and elevated apoptosis rate, causing dysfunctional neutrophils and SCN49,10.
Although SCN comprises a heterogeneous group of rare genetic diseases, SCN4 occurs mainly as a syndromic entity, including severe neutropenia and variable multisystemic non-hematologic features. In this respect, only seven cases of nonsyndromic neutropenia related to G6PC3 deficiency have been reported, all younger than 18 years when diagnosed4,11-13. Hence, the rarity of the disease, around one in 10 million people, and the identification of 11 patients suggestive of SCN4 in an adult tertiary medical center, prompted us to define the phenotypic, molecular, and geographical distribution characteristics of this apparent cluster in the Mexican population.
METHODS
Patient population
We conducted an observational retrospective study by reviewing the electronic medical records of patients with neutropenia (ICD-10 code D.70) seen from January 2000 to December 2020 at the National Institute of Medical Sciences and Nutrition “Salvador Zubiran” (Mexico City, Mexico). Syndromic neutropenia cases included at least one associated non-hematological disorder, such as IVSV, congenital heart defects, urogenital anomalies, and inflammatory bowel disease. Cases of secondary neutropenia due to other hematological or non-hematological disorders were excluded from the study.
Informed consent for study participation, molecular analysis, manuscript, and photographs publication is available from all the patients. The Institutional Review Board approved the study on May 21, 2021, (GEN-3729-21-22-1).
Clinical-genetic inclusion procedure
All patients included in the study were selected through a two-steps process by an experienced geneticist and a resident in medical genetics: (1) scrutiny from medical records of all cases with a diagnosis of neutropenia; and (2) a genetic assessment interview with each patient, including clinical exam, laboratory results, sociodemographic characteristics, pedigree construction, pre-test genetic counseling, signed consent approval and, from the patient and family members that accepted, a whole blood sample for the molecular study. Patients with phenotypic characteristics of other genetic or unrelated disorders were excluded from the study.
Molecular analysis
The DNA samples from all patients underwent direct Sanger sequencing of the whole coding region of the G6PC3 gene (RefSeq NM_138387.4). The primer sequences for G6PC3 (exon 1-6) were taken from a previous report2 and ordered from Integrated DNA Technologies (IDT, Coralville, Iowa, USA). We used HotStarTaq Master Mix Kit for fragment amplification under manufacturer standard conditions (QIAGEN, Hilden, Germany). The primer sequences and annealing temperatures are exhibited in Table S1. The PCR products were sequenced using a BigDye Terminator v3.1 Cycle Sequencing kit on an ABI Prism 3500 Genetic Analyzer under the procedure indicated by the manufacturer (Applied Biosystems, ThermoFisher Scientific, CA, USA). Sequence analysis was performed using the Unipro UGENE (version 39.0)14.
RESULTS
Patients
Eleven patients with neutropenia and non-hematologic traits were included in the study. The sex ratio female-to-male was 4:1. The current median age was 38.5 years (21-68 years); the median age at diagnosis was 32 years, although with a wide range (1 month-58 years). Regarding family backgrounds, patients 5 and 6 had first-degree relatives with recurrent infections, a sister and daughter, respectively, and patient 11 had two sisters with leukopenia.
Hematological findings
A varied hematological diagnosis was present in the four cases that were subsequently confirmed to have G6PC3 gene pathogenic variants (PV), showing neutropenia, cyclic neutropenia, pancytopenia, and medullary aplasia (Table 1 and Fig. 1). Of the remaining seven cases, two presented severe neutropenia and two cyclic neutropenia. Other diagnoses were mild neutropenia, cytopenia, and medullary aplasia, one each.
Table 1. Clinical description of patients with G6PC3 gene pathogenic variants
| Characteristics | P1 | P2 | P3 | P4 |
|---|---|---|---|---|
| Sex | Female | Male | Female | Male |
| Age | 25 | 37 | 28 | 19 |
| Consanguinity | Denied | Denied | Denied | Denied |
| ANC (cells/mm3) | 375–4051 | 84–20,942 | 297–4384 | 300–11,500 |
| Lymphocytes (cells/mm3) | 175–825* | 171–780* | 257–884* | 300*–5,814 |
| Platelets (×109/L) | 245-573 | 17†-257 | 91†-448 | 45†-499 |
| Height (cm) | 1.34 | 1.62 | 1.57 | 1.57 |
| Dysmorphic features | Triangular face, midface hypoplasia, depressed nasal bridge, thick lips, and prognathism | Round face, midface hypoplasia, depressed nasal bridge, thick lips, and prognathism | Round face, midface hypoplasia, depressed nasal bridge, thick lips, and prognathism | Round face, midface hypoplasia, thick lips prognathism |
| Congenital heart defects | ASD | ASD | ASD | ASD |
| Skeletal findings | Scoliosis | None | None | Pectus carinatum and scoliosis |
| Increased visibility of superficial veins pattern | Upper and lower limbs | Upper and lower limbs | Chest, upper and lower limbs | Absent |
| Endocrine findings | Delayed bone age, central thyroid dysfunction | Hypergonado-tropic hypogonadism | None | None |
| Bone marrow findings | Not available | Hypercellular marrow with myeloid hyperplasia | Increased megakaryocytes | Hypocellular marrow with megaloblastic changes in myeloid cell line |
| Other findings | Delayed psychomotor development, juvenile rheumatoid arthritis (JRA), and Crohn's disease | Crohn’s disease 46, XY |
Chronic diarrhea | Delayed psychomotor development 46, XY |
| Genotype | Compound heterozygous [c.210delC] + [c.199_218+1del] |
Homozygous [c.210delC] |
Homozygous [c.210delC] |
Homozygous [c.210delC] |
*Values in the range of lymphocytopenia; †Values in the range of thrombocytopenia; ASD: atrial septal defect.
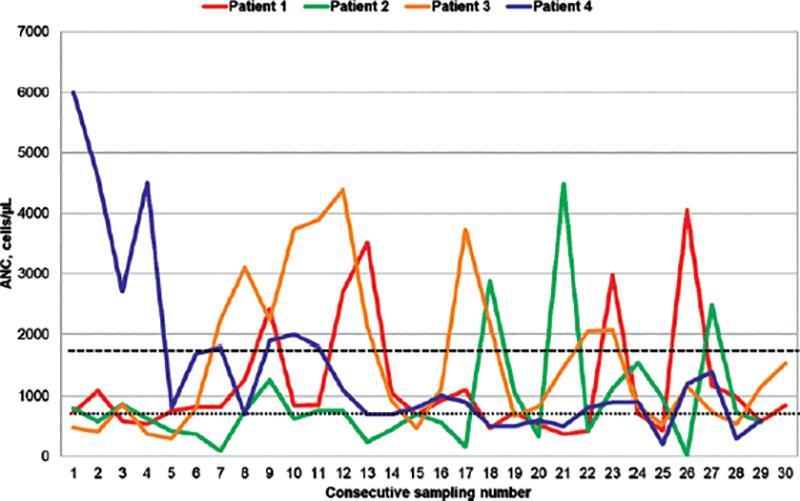
Figure 1. Absolute Neutrophilic Count (ANC) in G6PC3 deficient patients. Long dashes indicate the threshold of 1500 cells/uL, below which ANC corresponds to neutropenia; short dashes indicate the threshold of 500 cells/uL, corresponding to severe neutropenia. Spikes are coincident with remission episodes.
Non-hematological findings
The most frequent non-hematological findings in the patients were cardiovascular disorders in 8/11 cases (77.7%), atrial septal defects (ASD) in four patients, and a persistent cardiac murmur in the other 4. Furthermore, seven patients (63.6%) showed peripheral vascular system-associated symptoms, of which 6 had IVSV, and one showed peripheral venous insufficiency. In addition, four patients had anomalies of the urogenital tract (36.4%). A similar number of cases (36.4%) presented inflammatory bowel disease, Crohn’s disease in 2, and ulcerative colitis in one. Figure 2 shows the combined non-hematological manifestations of each of the 11 patients.

Molecular findings
Sanger sequencing of the G6PC3 gene confirmed the clinical diagnosis of SCN4 in 4 of the 11 patients analyzed (Fig. 3). Patient one was a compound heterozygous for the c.210delC variant (rs769441127; NC_000017.11:g.44071174) and a 21bp deletion c.199_218+1del (rs1597905369; NC_000017.11:g.44071159-44071179del), predicted to impact the canonical splice site region between exon and intron 1. The remaining three cases were homozygous for the frame shift pathogenic c.210delC variant creating a premature stop codon, just 46 amino acids after the deletion site, giving place to a truncated protein of 115 amino acid residues (NP_612396.1: p.(Phe71Serfs*46)) that presumed to suffer the nonsense-mediated decay of the transcript9. Segregation analysis was available for patients 1, 2, and 4. The 21bp deletion of compound heterozygous (patient 1) was inherited from the mother and the 1bp deletion from the father. Both parents of patients 2 and 4 were carriers of the c.210delC variant. Cascade testing in the family of patient 2 confirmed homozygosity for the c.210delC variant in 2 cousins of the index patient. One is a 24-year-old female with recurrent aphtha, cardiac murmur, and inguinal hernia, and her brother, 16 y. o., with recurrent upper airway infections and bilateral cryptorchidism. Although in the family of patient three, parents and siblings rejected genotyping, the mother and the father should be both heterozygous for the c.210delC variant because patient three is homozygous for it.

Figure 3. Electropherograms of the G6PC3 reference sequence
and the two identified pathogenic variants. Homozygous c.210delC in
patients 2, 3, and 4, and compound heterozygous c.210delC c.199_218+1del
variants in patient 1.
The first variant was inherited from the
father and the second from the mother.
Clinical description of positive cases
Table 1 compares the clinical findings observed in patients with a confirmed molecular diagnosis. Figure 1 shows the heterogeneous behavior of the magnitude of neutropenia over time in these four patients with the G6PC3 gene variants described.
Additional clinical findings observed in the cases with confirmed SCN4
Patient 1
Female, 25 y.o., born to an at-term uneventful pregnancy from healthy parents denying consanguinity. No other family member was affected (Fig. 4A). Soon after birth, a urinary infection required hospitalization for 4 days, solved without complications. The heart murmur, diagnosed at birth as an ASD and later developing to pulmonary arterial hypertension, was surgically repaired at age 14. She showed failure to thrive, delayed psychomotor development, recurrent infections during childhood with fever, cyclic neutropenia, and later, symptoms of juvenile rheumatoid arthritis. When first seen by us, she showed short stature of 134 cm (< 3rd centile), a head circumference of 49 cm (< 3rd centile), with an upper/lower segment ratio of 0.90 and arm span/height ratio of 0.94. She had a triangular face, retrognathia, mild frontal bossing, hypotelorism, low nasal bridge, short philtrum, downturned mouth corners, midface hypoplasia, IVSV of the upper and lower limbs, scoliosis, bilateral clinodactyly of the fifth finger, amenorrhea, central thyroid dysfunction, and growth hormone release defect. She was diagnosed with cyclic neutropenia and nonregenerative hypochromic microcytic anemia secondary to iron deficiency. A colon biopsy performed due to chronic diarrhea showed a focal zone of fibrosis, active inflammation in the terminal ileum, collapsed loops, and mesenteric adenomegaly diagnosed as Crohn’s disease.

Figure 4. Sequencing analysis results and genealogies of G6PC3 mutation positive. Family pedigrees illustrate the four cases in which a pathogenic variant (PV) in the G6PC3 gene was present. In addition, segregation analysis and cascade testing were done to determine the parental origin of variants and carriers in three families. Circles represent females; squares indicate males; arrows refer to probands; fully shaded figures represent SCN4 patients, symbols with a black dot inside represent PV carriers. *Family members who underwent genetic testing. Arabic numerals-individual identifiers; Roman numerals-generations.
Patient 2
Male, 37 y.o., born to an at-term uneventful pregnancy of probably consanguineous healthy parents from a rural community (933 inhabitants); and a family history of a female second cousin who died at age 18 of severe neutropenia and Crohn’s disease (Fig. 4B). At birth, he presented neutropenia, later diagnosed as cyclic neutropenia. He has bilateral cryptorchidism, surgically repaired at age 8. He has short stature (162 cm, p < 5), dysmorphic face features (Table 1), an IVSV pattern of the upper and lower limbs, acute kidney pain, kidney stones, and hypercalciuria. He presented intense-generalized abdominal pain and episodic diarrhea in late adolescence. A colon biopsy at age 19 showed acute severe ulcerative colitis and rectum with chronic proctitis diagnosed as Crohn’s disease, treated surgically, and currently having an ileostomy. Incidentally, the patient was diagnosed with ASD at the same age, which was repaired. Sex hormone analysis confirmed hypogonadotropic hypogonadism, with a 46, XY normal karyotype. He is currently on and off granulocyte colony-stimulating factor treatment.
Patient 3
Female, 28 y.o., born to an at-term uneventful pregnancy of unknown consanguinity of healthy parents (Fig. 4C). She presented cytopenia, recurrent otitis media, and upper airway infections during childhood. At age 19, she started with episodic diarrhea and intense-generalized abdominal pain, mild neutropenia, thrombocytopenia, and lymphopenia. On genetic evaluation, she presented dysmorphic features (Table 1), with upper and lower limbs showing IVSV. Echocardiography at 28 years of age confirmed an ASD and mitral and aortic insufficiency.
Patient 4
Male 19 y.o., born to an at-term uneventful pregnancy and ignored consanguinity of healthy parents (Fig. 4D). He had recurrent upper-airway infections, hypotonia, and gastroesophageal reflux during the neonatal period. Recurrent upper airway infections and idiopathic neutropenia continue as current symptoms. At age 16, he underwent surgical repair of a previously diagnosed ASD. Imaging studies confirmed thoracic 2-4 vertebral fusion and dextroscoliosis. He also presented dysmorphic features and Pectus carinatum (Table 1).
Interestingly, even though patients denied or ignored kinship and consanguinity, the information on geographic family origin from the father of patient 1, both parents of patient 2, and the father of patient 3 confirmed that they are all from neighboring towns of the same state (patient 1, Ixtlahuaca; patient 2, Jiquipilco; patient 3, Toluca; patient 4, Chalco). Furthermore, the distances between locations show geographical proximity: Ixtlahuaca-Jiquipilco 14 km, Ixtlahuaca-Toluca 34 km, Jiquipilco-Toluca 31 km, and Chalco-Toluca-Ixtlahuaca-Jiquipilco, 77–97 km. This strongly suggests a founder effect for the G6PC3 gene c.210delC variant. Moreover, sharing an uncommon surname by two of the families further supports the possibility of a common ancestor, probably for all families.
DISCUSSION
We conducted a phenotypic, biochemical, and genetic analysis in 11 patients with severe cyclic congenital neutropenia. Sanger sequencing identified two previously rare reported G6PC3 gene PVs in 4 of the 11 patients with non-hematologic traits (36.4%). Patient 1 had a rare splice donor site variant (c.199_218+1del), which has only once been submitted to the ClinVar database (ID: 691994), in a patient with pulmonary arterial hypertension, leukopenia, and an ASD, probably having SCN4, although not specified in the report15. The c.210delC variant has been well characterized as pathogenic and has only been described in patients of Hispanic descent5,16. Interestingly, 31 heterozygous carriers for the c.210delC variant are reported in the gnomeAD database, all in Latino/Admixed Americans17; in addition, 395 alleles were identified in 92,041 individuals (184,082 alleles) of Native Mexican ancestry, with a prevalence of 0.00215 in the Mexico City Prospective Study (MCPS) database18.
Furthermore, in the same database, this variant has the highest frequency of all PV of the G6PC3 gene, with a prevalence of 0.00143 in 138,200 DNA-sequenced samples. Interestingly, the c.210delC variant was only found in individuals confirmed to be of Native Mexican ancestry but not in Mexicans with an admixture of African, Asian or European ancestry18. Although our patients denied inter-family relationships, the family residences of patients 1, 2, and 3 are within a 35 km distance, which makes it possible that these families share a common ancestor, also supported by the fact that two of the families share an uncommon surname. In another study of Mexican patients, Velez-Tirado et al. reported 5 patients carrying this variant; 3 of them were homozygotes, and although they denied consanguinity, the authors mentioned that they might come from geographically close communities16. Our findings and the above reports strongly support that the c.210delC variant is particular to the Mexican population and sheds light on a possible founder effect5,16-18.
We describe four patients with sequencing diagnoses of G6PC3 gene PV, 3 of which are homozygous for the c.210delC nonsense variant. Although the disease is known as SCN4, the blood analysis findings in these patients show that the hematological phenotype is much broader. Furthermore, our patients (Table 1) fulfilled the criteria for SCN and cyclic neutropenia, as all showed variable degrees of severe neutropenia associated with periods of remission and affected platelet and lymphocytes cell lines. Regarding non-hematological abnormalities, congenital heart defects were the most frequent, as others have noticed16,19. We found that in all positive cases presented with ASD, these defects usually occurred as soft murmurs or were asymptomatic for a long time, making them a common undiagnosed abnormality20. In our group of patients, IVSV was a relatively common finding. Although patient four did not show this anomaly, veins may probably become more prominent with age5, being this finding present in two-thirds of patients16.
Regarding patients without G6PC3 PV, despite the unspecific diagnosis of a heart murmur, none of these patients had an ASD, as observed in the four patients with a G6PC3 PV, which has been frequently associated with mutations in this gene16. Although 3/7 had IVSV, in our sample of cases, it seems not to be a defining phenotypic marker of the syndromic SCN4. In addition, except for one patient with ulcerative colitis, no other patients showed gastrointestinal symptomatology. The common aspect in both groups was the heterogeneous character of hematologic manifestations. It is of note that SCNs are a group of heterogeneous diseases with different inheritance and multigene etiology1. Therefore, the syndromic form is probably not always exclusively due to mutations in the G6PC3 gene.
G6PC3 deficiency due to pathogenic and probably PV of the G6PC3 gene is considered within the group of rare diseases. The 127 reported cases of SCN4 worldwide present 53 different G6PC3 PV. Of these, 22 are missense (41.5%), 22 nonsense or frame shift (41.5%), 6 splice site variants (11.3%), and 3 in-frame deletions (5.7%); gene location and detailed characteristics of each one is shown in figure 5 and table 2. Most described cases are in the pediatric age group, and in most, a diagnosis has been reached via WES, meaning that clinical diagnosis is rarely suspected; therefore, it is possible that SCN4 may be underdiagnosed. The 4 patients reported herein reached adulthood without a diagnosis, until now. An early confirmed diagnosis can diminish complications such as Crohn’s disease, believed to be caused by intrinsic defects in hematopoietic cells, with inadequate control of gut microbiota, secondary to reduced survival and function of neutrophils, leading to a dysregulated inflammatory response; this association is not specific to G6PC3 deficiency21. There are more than 240 risk genes for IBD, including Crohn’s disease; however, the pathogenic mechanism of contribution is not well understood. IBD is an immune-mediated disorder, and several identified genes participate in regulating and maintaining the local immune response to viruses and bacteria. The particular genetic background may predispose to altered immunoglobulin production, enhancement of specific cytokine expression, and subsequent tissue inflammation22. Crohn’s disease or another IBD may not be influenced directly by G6PC3 mutations but is aggravated by neutropenia. In addition, well-timed allogeneic hematopoietic stem cell transplantation has been proven beneficial in the remission of inflammatory bowel disease symptomatology23.
Table 2. Compilation of G6PC3 mutations reported in cases with SCN4
| Exon | NM_138387.4 | NP_612396.1 | Short | MC | GRCh38 chr17 | dbSNP | ClinVar | Interpr. | MT | CADD | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.[130C>T] | p.[Pro44Ser] | P44S | MS | 44071095 | rs775224457 | 189781 | P | DC | 28 | 1-5 |
| 1 | c.[131C>T] | p.[Pro44Leu] | P44L | MS | 44071096 | rs762019955 | na | na | DC | 29 | 6-8 |
| 1 | c.[141C>G] | p.[Tyr47Ter] | Y47* | N | 44071106 | rs118203970 | 1039 | P | DC | 37 | 6,8,9 |
| 1 | c.[144C>A] | p.[Tyr48Ter] | Y48* | N | 44071109 | rs1194477276 | 632283 | likely-P | DC | 34 | 10,11 |
| 1 | c.[175T>C] | p.[Trp59Arg] | W59R | MS | 44071140 | rs752966267 | 853860 | US | DC | 32 | 8,12 |
| 1 | c.[190_210]delACCGAGTGGCTCAACCTCATC | p.[Thr64_Ile70del] | T64_I70del | IFD | 44071155- 44071175 | na | na | na | DC | na | 2,3 |
| 1 | c.[194A>C] | p.[Glu65Ala] | E65A | MS | 44071159 | rs745318917 | na | na | DC | 31 | 7,13 |
| 1 | c.[207delC] | p.[Ile70fsTer46] | I70fs* | FS | 44071172 | na | na | na | DC | na | 14 |
| 1 | c.[207dup] | p.[Ile70fsTer17] | I70fs* | FS | 44071171- 44071172 | rs1191239079 | 691992 | P | DC | 33 | 6 |
| 1 | c.[210delC] | p.[Phe71fsTer46] | F71fs | FS | 44071175 | rs769441127 | 189782 | P | DC | 35 | 6,15 This publication |
| 1 | c.[214delA] | p.[Lys72fsTer45] | K72fs* | FS | 44071179 | rs1177939839 | na | na | DC | na | 16 |
| 1 | c.[218+1G>A] | p.? | p.? | SDL | 44071184 | na | 1430600 | P | DC | 35 | 6,17 |
| 1 | c.199_218+1delCTCAACCTCATCTTCAAGTGG | p.? | p.? | SD | 44071159-44071179 | rs1597905369 | 691994 | P | DC | na | This publication |
| 2 | c.[249G>A] | p.Trp83Ter | W83* | N | 44074190 | na | na | na | DC | 39 | 18 |
| 2 | c.[257delA] | p.[Glu86Glyfs*31] | E86Gfs* | FS | 44074198 | na | na | na | DC | na | 19 |
| 2 | c.[282delA] | p.[Ala95fsTer22] | A95fs* | FS | 44074223 | na | na | na | DC | na | 5 |
| 2 | c.[295C>T] | p.[Gln199Ter] | Q99* | N | 44074236 | na | na | na | DC | 38 | 20 |
| 3 | c.[326-1G>C] | p.? | p.? | SAL | 44074679 | na | na | na | DC | 33 | 7 |
| 3 | c.[346A>G] | p.[Met116Val] | M116V | MS | 44074700 | rs267606834 | 1042 | P | DC | 25 | 21 |
| 3 | c.[347T>C] | p.[Met116Thr] | M116T | MS | 44074701 | na | na | na | DC | 26 | 1,5 |
| 3 | c.[347T>A] | p.[Met116Lys] | M116K | MS | 44074701 | na | na | na | DC | 28 | 22 |
| 3 | c.[348G>A] | p.[Met116Ile] | M116I | MS | 44074702 | rs1373865222 | na | na | DC | 30 | 6 |
| 3 | c.[353C>G] | p.[Thr118Arg] | T118R | MS | 44074707 | rs766706036 | na | na | DC | 25 | 14 |
| 3 | c.[372delC] | p.[Ile125*] | I125* | N | 44074726 | na | na | na | DC | na | 23 |
| 3 | c.[373_375delATA] | p.[Ile125del] | I125del | IFD | 44074727-44074729 | na | na | na | DC | na | 11 |
| 3 | c.[394C>T] | p.[Gln132Ter] | Q132* | N | 44074748 | na | na | na | DC | 36 | 7,24 |
| 3 | c.[416G>T] | p.[Ser139Ile] | S139I | MS | 44074770 | na | na | na | DC | 39 | 6,25 |
| 4 | c.[417-1G>A] | p.? | p.? | SAL | 44074968 | rs763408993 | na | na | DC | 34 | 26 |
| 4 | c.[421delT] | p.[Trp141fsTer2] | W141fs* | FS | 44074973 | na | na | na | DC | na | 15 |
| 4 | c.[461T>C] | p.[Leu154Pro] | L154P | MS | 44075013 | na | na | na | DC | 28 | 27 |
| 4 | c.[481C>T] | p.[Arg161Ter] | R161* | N | 44075033 | rs1056739194 | na | na | DC | 35 | 15,18 |
| 4 | c.[482G>A] | p.[Arg161Gln] | R161Q | MS | 44075034 | rs1485073209 | na | na | DC | 27 | 6 |
| 4 | c.[482G>C] | p.[Arg161Pro] | R161P | MS | 44075034 | na | 1679807 | US | DC | 26 | 28 |
| 4 | c.[535+1G>A] | p.? | p.? | SS | 44075088 | na | na | na | DC | 34 | 7,24 |
| 5 | c.[554T>C] | p.[Leu185Pro] | L185P | MS | 44075328 | rs118203969 | 1038 | P | DC | 24 | 15 |
| 5 | c.[565C>T] | p.[Arg189Ter] | R189* | N | 44075339 | rs745582203 | 653016 | P | DC | 36 | 6,8,18 |
| 5 | c.[566G>A] | p.[Arg189Gln] | R189Q | MS | 44075340 | rs140294222 | 262367 | US | DC | 18 | 22 |
| 5 | c.[596A>G] | p.[Tyr199Cys] | Y199C | MS | 44075370 | rs1597910284 | na | na | DC | 27 | 20 |
| 5 | c.[623T>G] | p.[Leu208Arg] | L208R | MS | 44075397 | na | na | na | 26 | 3 | |
| 5 | c.[677+1G>A] | p.? | p.? | SS | 44075452 | rs778208850 | 1066709 | likely-P | DC | 34 | 10 |
| 6 | c.[1000_1001delAT] | p.[Met334fs] | M334fs | FS | 44076002-44076003 | na | na | na | DC | na | 1 |
| 6 | c.[680_684delinsT] | p.[Ser227Leufs*3] | S227Lfs* | FS | 44075682-44075685 | na | na | na | DC | na | 11 |
| 6 | c.[757C>T] | p.[Arg253Cys] | R253C | MS | 44075759 | rs765927570 | 960968 | likely-P/US | DC | 31 | 29 |
| 6 | c.[758G>A] | p.[Arg253His] | R253H | MS | 44075760 | rs118203968 | 1037 | P | DC | 31 | 10,18,20,22,30 |
| 6 | c.[935dupT] | p.[Asn313fs] | N313fs+39aa | FS | 44075933-44075934 | rs797044567 | 189784 | P | DC | na | 6,7,10,26,27 |
| +38 AA | FS | ||||||||||
| 44075767 | rs748931188 | 1342134 | P | DC | 33 | 7,33-36 | |||||
| 6 | c.[766_768delGGG] | p.[Gly256del] | G256del | IFD | 44075768-44075770 | na | na | na | DC | na | 20 |
| 6 | c.[778G>C] | p.[Gly260Arg] | G260R | MS | 44075780 | rs200478425 | 30874 | P/US | DC | 28 | 6,10,14,18,22,26,33,34 |
| 6 | c.[779G>A] | p.[Gly260Asp] | G260D | MS | 44075781 | na | na | na | DC | 27 | 6 |
| 6 | c.[829C>T] | p.[Gln277Ter] | Q277* | N | 44075831 | rs148559256 | 189783 | P | DC | 33 | 6,10,35,36 |
| 6 | c.[882_903dup] | p.[His302GlyfsTer92] | H302Gfs* | FS | 44075884-44075905 | na | na | na | DC | na | 5 |
| 6 | c.[765_766delAG] | p.[Ala257CysfsTer129] | A257Cfs*+38aa | FS | 44075767 | rs748931188 | 1342134 | P | DC | 33 | 7,33-36 |
| +39 aa | FS | 44075933-44075934 | rs797044567 | 189784 | P | DC | na | 6,7,10,26,27 | |||
| 6 | c.[960delG] | p.[Trp320CysfsTer4] | W320Cfs* | FS | 44075961 | na | na | na | DC | na | 32 |
| 6 | c.[974T>G] | p.[Leu325Arg] | L325R | MS | 44075976 | na | na | na | DC | 25 | 37 |
MC: molecular consequence; MT: mutation Tester prediction; MS: missense; N: nonsense; FS: frameshift; IFD-in-frame deletion; SDL: splicing donor lost; SD: splicing donor; SAL: splicing acceptor lost; SS: splicing site; P: pathogenic; na: not available; US: uncertain significance; DC: disease causing; CADD: Combined Annotation Dependent Depletion score. †References mentioned in this table are placed in the Suplementary Data.

Figure 5. G6PC3 reported pathogenic variants (PV) in patients with SCN4. (A) the absolute frequency of unrelated patients (one per family) of known PV in confirmed SCN4 cases reported in the literature consulted in Scholar Google. (B) illustration of known G6PC3 PV presented on the protein with structural domains indicated. The red rectangle encloses a pathogenic variant found only in the Mexican population. All variant details and references can be found in Table II and Appendix S1, respectively.
It is known that SCN is a heterogeneous disease with multigene involvement and diverse inheritance patterns, as well as variable phenotypic manifestations, being ELANE (neutrophil elastase gene) PV responsible in most cases with the disease. However, such a form of SCN occurs without specific non-hematological manifestations1. Moreover, only two Mexican patients were reported with PV in ELANE and were sporadic24. Meanwhile, five identified SCN patients were found to have genetic alterations on G6PC316. Therefore, the characterization of non-hematologic SCN-associated multisystemic external and internal abnormalities in the patients in this study shed light on defining the SCN4 syndromic form of the disease.
The findings in the four patients, mainly those present in the three patients homozygous for the c.210delC variant, do not allow to propose a definitive genotype-phenotype correlation, even though patients share the same congenital heart defect and other phenotypic features, such as IVSV, gastrointestinal, and characteristic facial appearance. Regarding the rest of the systems involved, findings were heterogeneous, which may represent, to some extent, a variable expression of this pathogenic variant, although other yet unknown modifying factors may affect the occurrence of non-hematological manifestations.
Finally, the present uniqueness of the c.210delC variant in Hispanics of Mexican descent, the regional proximity of the families’ residence, and the sharing of an uncommon surname between the members of two of the families, strongly support the possibility of ignored consanguinity, an ancient common ancestor, and a founder effect for this G6PC3 gene variant.
Our study showed that searching for non-hematological features in patients with severe neutropenia could identify cases with G6PC3 PV. Therefore, it is highly recommended that patients with primary neutropenia be examined mainly for congenital heart defects, peripheral vascular system anomalies, inflammatory bowel disease, urogenital defects, and facial appearance, to guide genetic testing for decision-making. Furthermore, our findings suggest this disorder may have a higher prevalence in Mexicans than has been described, most probably due to a founder effect of the c.210delC pathogenic variant, particular to the Mexican population.
SUPPLEMENTARY DATA
Supplementary data are available at Revista de Investigación Clínica online (10.24875/RIC.22000234). These data are provided by the corresponding author and published online for the benefit of the reader. The contents of supplementary data are the sole responsibility of the authors.

