











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkTransthyretin (TTR) amyloidosis (ATTR amyloidosis) is a debilitating, progressive, and eventually a fatal condition characterized by multiorgan accumulation of protein-like material in a fibrillar shape identified as amyloid deposits. It was first described as familial amyloid polyneuropathy (FAP) in North Portugal, Japan, and Sweden1. Until 1990, it was considered to be a rare disease; however, more comprehensive application of advances in genetic tests enabled the identification of the disease globally. The Nomenclature Committee of the International Society of Amyloidosis in 2018 recommended using ATTR amyloidosis instead of FAP, emphasizing the systemic involvement of the condition and the amyloidogenic protein2.
Formerly known as pre-albumin, TTR is a plasma protein responsible for the transport of retinol (Vitamin A) and thyroxine. TTR is mainly synthesized and excreted by the liver, although the epithelial cells of the choroid plexus and the retinal pigment in the epithelium also locally express TTR. In the pathologic state, TTR forms amyloid fibril aggregates with aging (wild-type [ATTRwt] amyloidosis) or in the presence of missense mutations (ATTRv amyloidosis). Over 130 mutations of the single gene encoding TTR have been described. It is inherited in an autosomal dominant fashion. TTR is a native tetrameric structure that is more unstable and prone to dissociation into monomers in the presence of point mutations or aging. Protein aggregation is mainly due to the reduced folding stability of the monomers, resulting in insoluble amyloid fibrils capable of systemic extracellular deposition3,4. The amyloidogenic potential of TTR variants correlates inversely with their thermodynamic stability3. Amyloid deposits occur in the somatic and autonomic nervous system and heart, and less frequently in eyes, kidneys, leptomeningeal vessels, joints, and ligaments3. Endogenous or local chemophysical factors and biomechanical forces might promote tissue specificity. Amyloid fibrils may cause tissue damage by direct compression or obstruction, or by neurotoxicity provoked by non-fibrillar oligomers and protofibrils. Finally, blood vessel deposits may cause microangiopathy3.
Over the past decades and despite being a rare disease, ATTR amyloidosis has stimulated the scientific community interest, enabling top-tier therapeutics5. In the 90s, organ transplantation was the mainstream therapeutic option, and it fostered distinct approaches: patients were candidates to receive two to three organs in the same or consecutive procedures. Furthermore, it enabled strategies to overcome the shortage of organ donations by developing a domino liver transplant (DLT) strategy, allowing the utilization of the amylogenic liver of the younger patients in older adults with fatal liver failure6. This strategy provided the first step toward more sophisticated transplant chain donations.
Likewise, several oriented genetic therapies emerged to prevent, control, and, surprisingly, reverse amyloid deposition. ATTR amyloidosis is one of the first conditions that use different RNA silencing strategies: antisense oligonucleotides (ASAs) and small interfering RNA molecules.
The uniqueness of the transplant modalities and the novelty of genetic manipulation make this disorder a role model for developing other disease-specific therapies. The experience gained will boost the advancement of new genetic modifiers. Following the success of ATTR amyloidosis, it is possible that manipulating RNA variations will become a frequent strategy in the treatment, diagnosis, and prevention of many common and rare diseases. The development and implementation of therapy for ATTR amyloidosis over the past two decades (Fig. 1) and their application in other medical conditions are briefly discussed in this article.
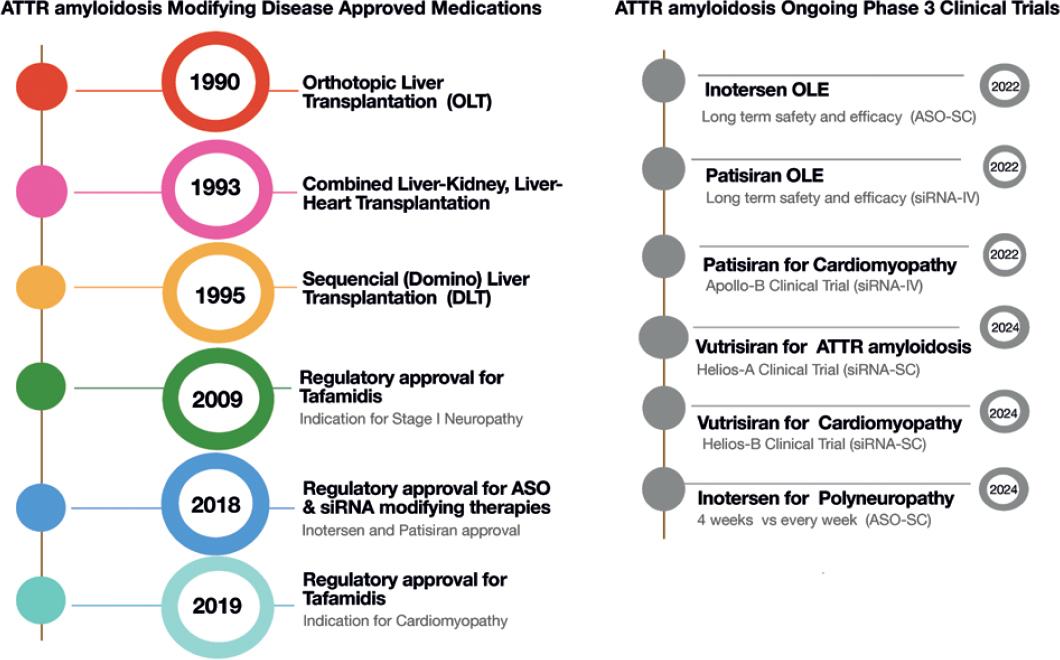
Figure 1 Development of transthyretin amyloidosis modifying treatments. OLE: open-label extension; ASO: anti-sense oligonucleotide; siRNA: short interference RNA; SC: subcutaneous; IV: intavenous.
ORGAN TRANSPLANTATION
Orthotopic liver transplantation (OLT) was the first disease-modifying treatment for ATTRv amyloidosis (Fig. 1). Introduced in 1990, OLT reduces the production of mutant TTR in the affected liver, the source of 98% of the body’s TTR. It is particularly beneficial in the early stages of the disease, preventing amyloid fiber deposition production and avoiding organ dysfunction. Newer modalities followed the classic OLT procedure (Fig. 1A) like combining heart and liver transplantation in patients with cardiac amyloid infiltration at diagnosis. This modality is performed with organs from the same or different donors and in the same or successive procedures. Both organ transplants share the indications for immune suppression, so the morbidity does not increase after the procedure. Other combined modalities include renal or corneal transplants.
In the mid-1990s, DLT became the preferred modality in these patients7. DLT comprises transplanting the liver from a patient with ATTRv amyloidosis into a patient with end-stage liver disease. The logic behind this procedure is that the recipient is too sick to wait for another (healthier) liver and old enough to have a life free of amyloidosis, despite receiving a liver with mutations. Furthermore, the amyloid patient is placed on top of the list despite not having end-stage liver failure. The procedure helps the organ shortage and eases the waiting list for receiving a transplant. A careful selection of the recipients must be done7 (Fig. 2A). This modality is now being tested in maple syrup urine disease, acute intermittent porphyria, hereditary fibrinogen A chain amyloidosis, and many other non-cirrhotic inherited metabolic liver diseases that have anatomically and functionally normal livers except for the enzymatic defect. Not all liver enzyme conditions have a good outcome with these procedures, and other factors, including non-hepatic enzyme activity to compensate for the metabolically deficient graft, have to be explored. Alternative therapeutic approaches to OLT are also vigorously explored, such as hepatocyte transplantation, combining different organs with distinct metabolic diseases, or combining a native and non-cirrhotic liver with metabolic defects. These procedures are currently changing the rule book of liver transplantation in a world with scarcely available organ donations.
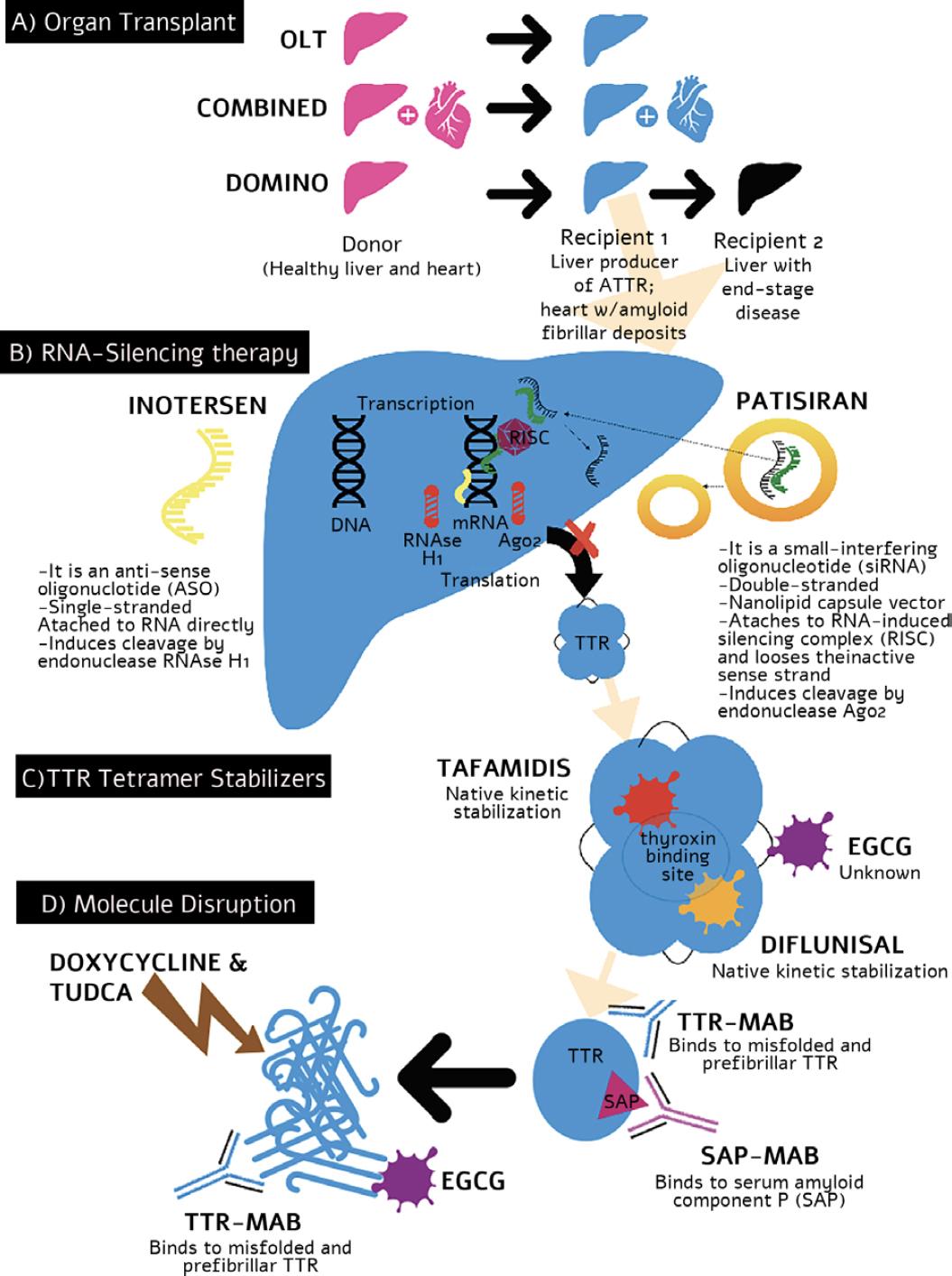
Figure 2 Mechanisms of the action of transthyretin amyloidosis modifying treatments. OLT: orthotopic liver transplant; MAB: mononuclear antibody; EGCG: epigallocatechin-3-gallate; RISC: RNA-induced silencing complex; AGO: Argonaute 2 endonuclease; SAP: serum amyloid protein; ASO: anti-sense oligonucleotide; TUDCA: taurodeoxycholic acid.
OLT and its variants in ATTRv amyloidosis are becoming less popular as newer medications emerge with fewer side effects. Transplants carry risks, particularly infections and organ rejection. Furthermore, in some patients with preexisting ATTR cardiomyopathy, the cardiac amyloid deposition continues because of the toxic effect exerted by the already deposited amyloid7. Likewise, the production of mutant TTR in the choroid plexus and retinal pigmented epithelium continues. Patient eligibility, organ availability, and the risks of the procedure itself are also drawbacks. Moreover, some patients’ quality of life does not improve after transplantation because the need of multiple daily medications, or because they suffer a constant threat of organ rejection, and may have many follow-up medical appointments in several specialties.
TTR STABILIZERS
The first specific drug approved for ATTRv amyloidosis was tafamidis, a small molecule that prevents the serum TTR from dissociating in monomers and forming fibrillar amyloid (Fig. 1C). Tafamidis can reduce the progression of neuropathy and cardiomyopathy. Unfortunately, it does not stop the disease progression in all the ATTRv amyloidosis variants or in patients with advanced disease7. However, survival in patients treated with tafamidis is still longer than in untreated patients, which is around 10 years. Diflunisal is a non-steroidal anti-inflammatory drug that has not been approved for ATTRv amyloidosis; therefore, it is used “off-label.” It represents an alternative option in the places where it is available for patients without access to other treatment modalities. The mechanism of action is similar to tafamidis, with the advantage of having a meager cost compared to the other approved drugs. Erosive gastritis and gastrointestinal bleeding are the main side effects. Acoramidis is a potent kinetic stabilizer of TTR that was safe and effective in patients with cardiomyopathy, and two clinical trials are ongoing in subjects with cardiomyopathy and polyneuropathy8. Green tea contains epigallocatechin-3-gallate (EGCG), which induces a native state kinetic stabilization of TTR and can disaggregate amyloid deposits and disrupt preformed fibrils. The main caveat is that it interacts with human serum albumin and precludes the use of other more potent stabilizers such as tafamidis5. The use of stabilizers seems to be promising therapeutic options for asymptomatic or carrier patients. The future will include clinical trials with different treatment combinations and dosages. Their good tolerability and absence of side effects point toward a long-term treatment in very young asymptomatic carriers.
mRNA THERAPIES
In ATTR amyloidosis, there are approved and commercially available products for inactivating or “knocking out” the mutated TTR gene. Two different mechanisms exist, small interfering RNA (siRNA) and ASAs (Fig. 2B). siRNAs are a class of double-stranded RNA (dsRNA) molecules used to regulate the expression of genes by the mechanism of RNA interference (RNAi)9. A dsRNA is hybridized with a specific mRNA sequence to induce silencing or degradation of the TTR mutated expression. On delivery into cells, siRNAs bind to a multiprotein component complex, known as the RNA-induced silencing complex (RISC) in the cytoplasm; the two strands separate, and the strand with the RISC hybridizes with the target mRNA. After that step, argonaute-2 (Ago2), a catalytic component of the RISC, drives mRNA cleavage10, hindering the production of TTR. In 2018, the FDA approved patisiran, the first siRNA-based drug to treat hereditary TTR-mediated amyloidosis (hATTR). Patisiran is a double-stranded siRNA formulated as a lipid nanoparticle for targeted delivery to hepatocytes. In the Phase III clinical trial Apollo, IV infusions with patisiran significantly changed the primary and secondary outcomes compared with placebo11 and in the open-label extension12. A new formulation, revusiran, was designed to allow subcutaneous administration using a GAlNAC-conjugate nanovector instead of lipid nanoparticles13. Vutrisiran, an enhanced compound, is currently being tested in multicenter Phase III clinical trials to treat ATTR amyloid cardiomyopathy and neuropathy.
Inotersen is a 2’-MOE-modified antisense oligonucleotide (ASO)14. ASOs use short synthetic single-stranded DNA molecules complementary to specific pre-mRNA sequences to alter the splicing process14,15. Inotersen, in the Phase III clinical trial NEURO-TTR, was shown to stabilize or improve neuropathy symptoms and reduce TTR levels, but it did not change the cardiological outcomes significantly15,16. Current ongoing clinical trials evaluate long-term safety and efficacy and longer intervals between applications. An indirect comparison of the effectiveness of patisiran and inotersen through the published results of the main clinical trials showed that patisiran had significantly better treatment effects than inotersen on neuropathy and QOL5,17.
Transient knockdown of a defective gene causes decreased expression of the gene product, reducing the risk of the pathological state secondary to the mutation. In addition to the siRNA or ASOs, several different modalities are being developed, for instance, RNAse-H-dependent antisense, pre-mRNA splicing sites, or nuclease cleavage sites. The development of nucleic acid analogs that bind distinct targets is investigated to treat various conditions, including hemophilia A and B, hypercholesterolemia, HCV infection, or breast cancer, among many others. Moving beyond the liver to target other tissues and organs such as the skin, mucosa, or eyes will also have the advantage of easier administration, low-dose requirements, and higher affinity to surface receptors. The lessons learned with this disease guided the entrance to a new class of drugs. ATTR amyloidosis vanguard therapies lead to a rapid expansion of clinical trials with knock-out gene therapies.
MONOCLONAL ANTIBODIES
Antibodies against human serum amyloid P component (SAP) produced a potent, complement-dependent macrophage-derived giant cell reaction in mice, resulting in removal of the visceral amyloid deposits1. Monoclonal antibodies can also promote amyloid deposit removal and degradation and may be helpful as a diagnostic tool18. Dezamizumab, an anti-SAP IgG1 antibody, demonstrated safety and efficacy in a Phase I clinical trial, but its development was discontinued1. Other compounds may be helpful, although there are no ongoing human clinical trials at the moment18 (Fig. 2D).
DOXYCYCLINE AND TAURODESOXYCHOLIC ACID (TUDCA)
Doxycycline, a derivative of tetracycline, is an antibiotic, and TUDCA is a bile acid. They are both amyloid matrix solvents and are investigated to promote the removal of TTR deposits and repair the remaining tissue. They have a synergistic effect and work by lowering both fibrillar and non-fibrillar deposits. A Phase II clinical trial with doxycycline and TUDCA showed that this combination stabilizes the disease in patients with ATTRv amyloidosis with good tolerability and few side effects19.
CONCLUSIONS
ATTRv amyloidosis is a genetic disease that has served as a prototype for developing different novel treatment modalities. It was a model for expanding new transplant options, such as domino liver transplantation and combined organ transplants. TTR stabilizers were the next generation of modifying treatments and resulted in a slower progression of the disease in patients with hereditary and wild-type ATTR amyloidosis. In the past decade, silencing genes to arrest the production of the mutated protein showed to be very successful and safe. This latter approach has become a model to target other genetically induced degenerative, metabolic, or neoplastic conditions. Finally, siRNA delivery through specialized non-viral lipid nanocarriers is a more accessible and inexpensive mechanism to deliver the genetic oligonucleotides into the genes, opening a new era of targeted treatments.
Given the number of different therapeutic options for ATTR amyloidosis, it is evident that patients do not need to manifest disease progression to initiate treatment. Future perspectives need to address the optimal time for beginning and combining different medications not only to halt the symptomatic progression of the disease but also to inhibit all amyloid depositions permanently. However, some ethical and practical dilemmas need to be resolved, including the optimal time of genetic testing and the development of treatments with longer intervals of application or more permanent results.

