











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCTION
Fukuyama congenital muscular dystrophy (FCMD) is the most common form of a group of autosomal recessive disorders characterized by altered α-dystroglycan glycosylation and caused by FKTN gene mutations. However, FKTN gene mutations may cause a broad spectrum of clinical manifestations. The most severe phenotype is Walker-Warburg syndrome (WWS), characterized by type 2 lissencephaly, cerebellar malformation, hydrocephalus, retinal and anterior eye chamber malformations, and congenital muscular dystrophy. Most affected infants die during the 1st year of life1. FCMD is also a severe phenotype with severe congenital muscle wasting, structural brain alterations, cognitive impairment, and epilepsy. The clinical course is helplessly progressive, and death occurs at an average age of 16 years2,3. Limb-girdle muscular dystrophy without cognitive impairment is milder4, and cardiomyopathy with no or minimal skeletal muscle weakness is the mildest phenotype5,6.
The FKTN gene locus is 9q31, it has 10 exons and encodes a glycosyltransferase active in the Golgi complex called fukutin, involved in dystroglycan glycosilation7. FKTN mRNA expression patterns in the human brain suggest the protein may influence neuronal migration8,9. Fukutin is expressed ubiquitously in human tissues, although many aspects of its function in various tissues remain unknown10,11.
FCMD is the second most frequent form of childhood muscular dystrophy in Japan. Most Japanese patients are homozygous for a founder mutation consisting of a 3kb insertion into the 3 untranslated region of the FKTN gene12. Clinically, these patients have early disease onset, hypotonia, generalized muscle weakness, structural brain malformations, and cognitive impairment13. The first non-Japanese patient with a FKTN mutation was reported by Silan et al.14, a Turkish infant born to consanguineous parents with macrocephaly, bilateral buphthalmos, cataracts, hypotonia, and dyspnea, who died 10 days after birth. Molecular analysis revealed a homozygous 1bp insertion within exon five of the FKTN gene, causing a frameshift and a premature stop codon at amino acid position 157. Shortly after, another Turkish infant with Walker-Warburg syndrome born to consanguineous parents was found to be homozygous for a nonsense FKTN gene mutation (c.345_346GC; p.Gln116Ter)15. He had hypotonia, bilateral corneal clouding, severe hydrocephalus, brain stem hypoplasia, and absent corpus callosum. An increasing number of FKTN mutations have recently been reported in non-Japanese patients, with phenotypes ranging from WWS, muscle-brain-eye disease, FCMD, and at the mild end of the spectrum, limb-girdle muscular dystrophy with no mental retardation and cardiomyopathy with no or minimal skeletal muscle weakness16,17.
Our purpose was to describe two Mexican siblings with dilated cardiomyopathy (DCM) and no signs of skeletal muscle weakness or atrophy, found to be homozygous for a very low-frequency FKTN missense variant.
MATERIALS AND METHODS
Patients
We studied two Mexican male siblings who died from heart failure and DCM at ages 20 and 21 years. Their parents were consanguineous (first cousins) and had two other apparently healthy offspring (Fig. 1). The index case (IV-3) had a normal childhood with normal developmental milestones. He was first referred to the National Institute of Cardiology in Mexico City at age 17 years due to progressive exercise intolerance, dyspnea, and inferior limb edema. Physical examination showed jugular venous distention, hepatojugular reflex, mitral and tricuspid regurgitant murmurs, hepatomegaly, and ascites. Echocardiography revealed severe mitral and tricuspid regurgitation, as well as biventricular enlargement and dysfunction. Cardiac magnetic resonance (CMR) showed enlargement of right and left cavities (left ventricle end-diastolic and systolic volumes were 216 ml and 182 ml, respectively), biventricular dysfunction, and myocardial fibrosis (Fig. 2). Left ventricle ejection fraction (LVEF) and right ventricle ejection fraction (RVEF) were reduced (16% and 15%, respectively). Pharmacological treatment initially caused clinical improvement; however, 6 months later, the patient developed acute heart failure, renal failure, and hepatic congestion. Treatment with diuretics and intravenous inotropes led to temporary improvement, but episodes of decompensated heart failure continued. He was admitted 4 months later with cardiogenic shock and died at age 21 years.

Figure 1 Family pedigree. DNA samples were available only for individuals III-1, III-2, IV-2, IV-3, and IV-4.

Figure 2 Cardiac magnetic resonance of the index case and younger sibling. Four chamber view (Panel A) and short-axis view (Panel B) of the index case showing enlargement of both right and left cavities. Late gadolinium intramyocardial enhancement was found in the four chamber (Panel E) and short-axis view (Panel F), compatible with non-ischemic myocardial fibrosis. Four chamber view (Panel C) and short-axis view (Panel D) of the younger sibling showing very similar changes, also with non-ischemic myocardial fibrosis in the four chambers (Panel E) and short-axis view (Panel F). RA: right atrium; RV: right ventricle; LA: left atrium; LV: left ventricle.
The youngest sibling (IV-4) had a similar medical history, with an apparently healthy childhood and adolescence. At age 18 years, he developed progressive dyspnea. CMR showed enlargement of both right and left cavities, biventricular dysfunction, reduced LVEF and RVEF (13% and 27%, respectively), and diffuse fibrosis of septal predominance (Fig. 2). While receiving pharmacological treatment for heart failure, he died of sudden cardiac death at age 20 years. He had no clinical signs of muscular dystrophy, and unfortunately, no muscle biopsies were available to study α-dystroglycan glycosylation.
Methods
High-quality DNA samples previously extracted from blood of affected siblings, both parents and one unaffected sibling, were available for analysis. Targeted next-generation sequencing (NGS) was performed using TruSight-Cardio sequencing kit (Illumina) in a MiSeq System (Illumina). Post-run sequencing quality was assessed with FastQC (Babraham Bioinformatics, UK). Sequence reads were aligned with BWA Enrichment v2.1.0 and variant calling was performed using GATK v4.0. Variants were annotated with ANNOVAR (wannovar.wglab.org) and variant effect predictor (grch37.ensembl.org/Homo_sapiens/Tools/VEP). In silico programs included in ANNOVAR annotation were used to predict functional consequences of the missense change and MaxEntScan (hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) was used to predict splice site alterations. Variants of interest found in DCM-related genes were classified according to the American College of Medical Genetics criteria18 as benign, likely benign, of unknown clinical significance (VUS), likely pathogenic (LP) or pathogenic (P), with the help of Varsome19. Variants of interest were confirmed by capillary sequencing and were screened in all first-degree relatives.
The study was approved by the Comité de Ética en Investigación (Ethics Committee) of the Instituto Nacional de Medicina Genómica and the Instituto Nacional de Cardiología Ignacio Chávez, Mexico City, Mexico. All the family members participating in the study provided informed consent.
RESULTS
NGS of the index case (IV-3) revealed a homozygous mutation in the FKTN gene: NM_001198963.2: c.1270G>A p.Gly424Ser (rs752358445), causing a substitution of glycine (a non-polar aliphatic amino acid) for serine (a polar hydrophilic amino acid) at position 424 within the catalytic domain of the fukutin protein. Capillary sequencing confirmed the index case and his affected sibling was homozygous for this mutation, while both parents and an unaffected sibling (IV-2) were found to be asymptomatic FKTN Gly424Ser heterozygous carriers (Fig. 3). No other family members were available for molecular analysis.
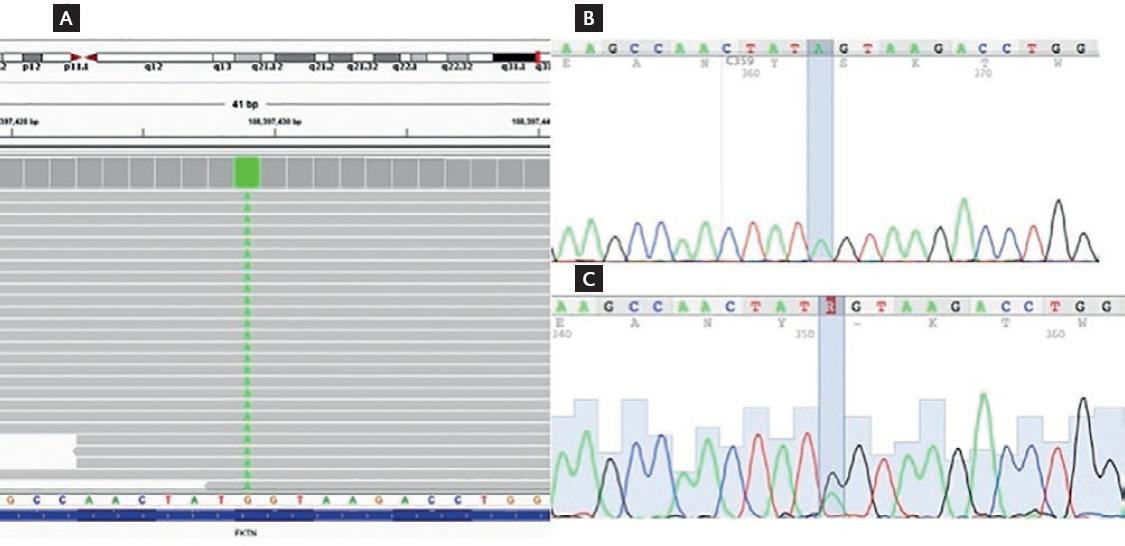
Figure 3 FKTN c.1270G>A (Gly424Ser) mutation. (A) Integrated Genome Viewer screenshot of the homozygous c.1270G>A mutation found in the index case. (B) Electropherogram confirming the homozygous c.1270G>A mutation, also found in the affected sibling (IV-4). (C) Electropherogram showing the heterozygous c.1270G>A mutation found in both parents and an unaffected sibling.
In silico programs, SIFT, Polyphen2, LRT, Mutation Taster, Provean, M-CAP, and Fathmm-MKL predicted the functional consequence of the amino acid substitution as damaging, while only two in silico programs (MetSVM and FATHMM) predicted the change as tolerated. Interestingly, in addition to the amino acid substitution, a loss of function splice site alteration was predicted by MaxEntScan for 3 of the 9 FKTN transcripts (ENST00000357998.5, ENST00000448551.2, and ENST00000457847.1).
The Gly424Ser substitution was classified as likely pathogenic, as the following AMCG criteria were met: moderate evidence of pathogenicity 2 (PM2: absent from controls or at extremely low frequency if recessive in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium), and supporting evidence of pathogenicity 1, 2, 3, and 4 (PP1: cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease; PP2: missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease; PP3: multiple lines of computational evidence support a deleterious effect on the gene or gene product; and PP4: patients phenotype or family history is highly specific for a disease with a single genetic etiology). Considering the possibility of a splice-site alteration, the mutation would also be classified as likely pathogenic.
DISCUSSION
The clinical findings of the siblings described here differ from FCMD, which is characterized by severe muscle wasting, hypotonia, and cognitive impairment. Our patients suffered severe DCM, rapidly progressive heart failure, and died at young age. However, because they showed no apparent signs of skeletal muscle weakness or neurologic symptoms, FKTN mutations were not suspected and muscle biopsies were not performed. Notably, two unrelated DCM patients without neurological symptoms who developed muscle weakness during adulthood were previously described. They were compound heterozygous for the FKTN 3kb insertion and one of two FKTN missense variants (p.Q358P and p.R179T)6.
In 2009, Arimura et al.17 further explored FKTN mutations in a group of 172 patients with DCM. The authors found a patient with mild muscular dystrophy and hyperCKemia but no brain involvement. This patient was compound heterozygous for FKTN mutations, the 3-kb insertion, and a missense Cys101Phe mutation. In addition, two DCM patients and three control subjects were heterozygous for the insertion and a normal FKTN allele, indicating that heterozygosity for the insertion itself is not associated with DCM. These observations suggest that compound heterozygous FKTN mutations could be a rare cause of DCM.
Nonsense and missense FKTN mutations may cause major damage to the fukutin protein and produce severe phenotypes. On the other hand, although the molecular mechanisms of FKTN mutations associated with DCM are unclear, fukutin function may be partially preserved in skeletal muscle of patients with minimal or late-onset muscle weakness. Further analyses are needed to clarify the function of fukutin in the glycosylation of α-DG in different organs6,2,10,11,17.
The FKTN Gly424Ser variant was found in only one Latino individual in the GnomAD database (https://gnomad.broadinstitute.org/) and is reported as of VUS in ClinVar (www.ncbi.nlm.nih.gov/clinvar/). However, because screening of first-degree relatives in this family revealed that both affected siblings were homozygous, and heterozygous carriers were asymptomatic (supporting evidence of pathogenicity), the variant met AMCG criteria to be classified as likely pathogenic. Moreover, while ClinVar mentions the WWS as a possible phenotype, it is known that homozygous loss of function FKTN mutations cause severe phenotypes, but compound heterozygous and missense mutations may cause milder phenotypes. Isolated DCM is considered as the mildest phenotype caused by FKTN mutations, and because our patients died at young age, it is unknown whether they may have developed skeletal muscle weakness at a more advanced age.
This is the first report demonstrating that heterozygous individuals for the c.1270G>A p.Gly424Ser mutation in the FKTN gene were healthy, while two homozygous brothers presented severe DCM, strongly suggesting that this mutation is a rare cause of autosomal recessive DCM.

