











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCTION
Immune thrombocytopenia (ITP) is an autoimmune disease characterized by increased platelet consumption and reduced production with platelet counts <100 × 109/L caused by both antibody and cell-mediated immune dysregulation1,2. ITP can be classified as primary, when no associated disease can be found, or secondary when ITP etiology is attributed to an underlying condition, including lymphoproliferative disorders, systemic lupus erythematosus (SLE), thyroid disease, antiphospholipid syndrome (APS), drug treatments, and infections such as human immunodeficiency virus (HIV), Helicobacter pylori infection, and hepatitis C or B virus (HCV, HBV)3. Although primary and secondary ITP share the generation of antiplatelet antibodies, they differ in specific aspects of pathobiology, natural history, and responsiveness to therapy4.
Secondary ITP diagnosis can be challenging due to the need to exclude the extensive differential diagnosis of thrombocytopenia and the lack of a specific diagnostic test. Recommendations for the initial diagnostic approach include patient history, physical examination, complete blood count, and peripheral blood smear examination. In addition, most authors routinely test adults for HCV, HBV, HIV, and, in appropriate geographical areas, H. pylori infection5. Autoimmune markers such as antinuclear antibody, rheumatoid factor, anticardiolipin antibodies, antithyroid peroxidase antibodies, and lupus anticoagulant have a higher prevalence in patients with ITP with no clinical evidence of these disorders; hence, they should not be routinely tested in the absence of disease-specific symptoms, although their presence can be the harbinger of autoimmune disease6. In addition, antiplatelet antibodies are only detected in 50-60% of patients and bone marrow examination is not diagnostic; thus, they are not recommended as an initial approach5-7.
Patients with primary or secondary ITP are often treated similarly. However, the current paradigm for secondary ITP may require treatment of the underlying disorder before treating thrombocytopenia3,5. Management might be difficult since corticosteroids and splenectomy are less effective than in primary ITP3. Establishing the diagnosis of secondary ITP requires precise targeting of the underlying disease, generally requiring an extensive clinical and laboratory workup.
We aimed to determine the incidence of secondary ITP over a decade at a referral hematology center, describe the underlying diseases, treatment modalities, long-term clinical outcomes, and chronic course predictors.
METHODS
This retrospective study included 258 consecutive patients diagnosed with ITP during the years 2008-2019. An underlying disease associated with secondary ITP was found in 83 (32%) patients. All patients had complete information in their clinical records and electronic files and were treated at the Department of Hematology of the Dr. José Eleuterio González University Hospital and the School of Medicine of the Universidad Autónoma de Nuevo Leon in Monterrey, Mexico. The study was approved by the Ethics and Research Committees of the institution and was in full compliance with the principles of the Declaration of Helsinki as revised in 2013.
Patients were divided into children <16 years and adults ≥16 years. The diagnosis was made according to clinical signs of bleeding and a platelet count <100 × 109/L.
Classification of disease stages was made following standard terminology, including newly diagnosed ITP when time between clinical symptoms and diagnosis was <3 months; persistent ITP from 3 to 12 months since diagnosis; and chronic ITP when the disease lasted ≥12 months. Diagnosis of severe ITP required the presence of clinically important bleeding manifestations5.
Initial treatment was selected by the treating physician according to the patients clinical history, comorbidities, and bleeding severity. Responses were classified as complete response (CR) when platelets were ≥100 × 109/L without bleeding manifestations and response (R) if there was a platelet count ≥30 × 109/L or a 2-fold increment from diagnosis without bleeding. No response (NR) was established by a platelet count <30 × 109/L, a <2-fold increase of the baseline platelet count or bleeding. Loss of response was defined as a platelet count <100 × 109/L or less than a 2-fold increase of the baseline platelet count8.
For the purposes of this analysis, children in whom a viral illness was documented during the 2 weeks preceding the beginning of ITP symptoms were classified as having secondary ITP4,9,10. Primary disorders in adults were divided into three groups: infections, SLE, and miscellaneous.
Statistical analysis
The statistical analysis was carried out with SPSS v.22 (IBM SPSS Statistics software, IBM Corp., Armonk, NY). Categorical variables are displayed as absolute numbers and percentages and comparisons were made with the Pearson x2 test. Quantitative variables were analyzed with descriptive statistics, including median and ranges. The MannWhitney U test was used for comparisons between quantitative variables. Risk factors for chronicity were studied by logistic regression analysis with a 95% confidence interval (CI). A p < 0.05 was considered statistically significant.
RESULTS
There were 83 patients with secondary ITP, 37 (44.5%) children, 46 (55.5%) adults; there were 52 (62.6%) women and 31 (37.4%) men, with a female to male ratio of 1.67:1 and a median follow-up of 21 (1-157) months. Overall response (OR) to first-line treatment was documented in 78 (93.97%) patients.
Pediatric Patients
Thirty-seven children younger than 16 years diagnosed with secondary ITP were included. There were 22 (59.5%) girls and 15 (40.5%) boys. Children had a median age of 3 (1-16) years with a median WHO bleeding scale of 1 (0-3). The main clinical characteristics are included in Table 1. Associated diseases in this group were previous infection in 32 (86.4%) patients; 25 were associated with respiratory tract infection, 5 with varicella-zoster virus and 2 with gastrointestinal infection; secondary ITP (SLE-associated) was confirmed in 4 (10.8%) children, 3 girls and 1 boy; and 1 boy (2.7%) had Evans syndrome.
Table 1 Clinical characteristics and laboratory features of 83 patients with secondary ITP treated at a University Hospital in Northeast Mexico
| Variable | Children | Adults |
|---|---|---|
| Total | 37 | 46 |
| Gender (%) | ||
| Male | 15 (40.5) | 16 (34.8) |
| Female | 22 (59.5) | 30 (65.2) |
| Age, years, median (range) | 3 (0-16) | 37 (17-73) |
| Clinical characteristics (%) | ||
| Petechiae | 26 (70.3) | 21 (45.7) |
| Ecchymosis | 25 (67.6) | 21 (45.7) |
| Gingivorrhagia | 4 (10.8) | 18 (39.1) |
| Epistaxis | 12 (32.4) | 11 (23.9) |
| Hematuria | 3 (8.1) | 3 (6.5) |
| Hematemesis | 1 (2.7) | |
| Lower GI bleeding | 1 (2.7) | 3 (6.5) |
| Metrorrhagia | | 6 (13.3) |
| Hemorrhagic bullae | 1 (2.7) | 2 (4.3) |
| Subconjunctival hemorrhage | | 2 (4.3) |
| Laboratory features, median (range) | ||
| Platelets × 109/L | 8.14 (0.257-98) | 13.1 (0.421-81) |
| Leukocytes × 109/L | 5 (4.69-16.1) | 8.34 (2.98-25) |
| Neutrophils × 109/L | 3.11 (0-27.6) | 5.84 (1.12-62) |
| Lymphocytes × 109/L | 3.03 (0-65.9) | 1.94 (0.55-52.1) |
| Chronic course (%) | 7 (18.9) | 14 (30.4) |
ITP: immune thrombocytopenia
Of the 37 children, 13 (35.1%) were treated exclusively with steroids, 12 (32.4%) with a combination of intravenous immunoglobulin G (IVIG) plus steroids, 8 (21.6%) received IVIG alone, 2 (5.4%) danazol plus steroids, and 2 (5.4%) children were under clinical observation. The initial response included 18 (48.6%) CR and 18 (48.6%) R; 1 (2.7%) was a non-responder diagnosed with secondary ITP (varicella-zoster virus-associated). The median time to maximum response was 7 (2-31) days. Responses to each treatment are summarized in Table 2. One patient presented neutropenia after treatment with steroids plus IVIG.
Table 2 Initial response in 37 children with secondary ITP receiving treatment at a reference center in Mexico
| Type of treatment | Complete response, n (%) | Response, n (%) | No-Response, n (%) |
|---|---|---|---|
| Steroids alone | 8 (61.5) | 4 (30.7) | 1 (7.6) |
| IVIG alone | 3 (37.5) | 5 (62.5) | |
| IVIG plus steroids | 6 (50) | 6 (50) | |
| Danazol plus steroids | | 2 (100) | |
| Clinical Observation | 1 (50) | 1 (50) | |
ITP: immune thrombocytopenia; IVIG: intravenous immunoglobulin G.
Relapse was documented in 7 (19.4%) children at a median of 15 (1-55) months. Six patients (85.7%) were treated again with steroids, and one (14.3%) with IVIG plus steroids; all achieved response. At last follow-up, 5 (13.5%) children had persistent disease and 7 (18.9%) followed a chronic course; of this last group, 4 (57%) were infection-associated and 3 (43%) SLE-associated. The median follow-up for children was 12.0 (range: 1-76) months.
Risk factors for chronicity were analyzed by logistic regression with a 95% CI. Uni- and multi-variate analysis results are shown in Table S1; age ≥10 years and a platelet count >20 × 109/L at diagnosis were statistically significant.
Adults
Secondary ITP diagnosis was established in 46 adults, 30 (65.2%) women and 16 (34.8%) men for a female to male rate of 1.9:1. A median age of 37 (17-73) years with a median WHO bleeding scale of 2 (0-3) was found, clinical characteristics at diagnosis are shown in Table 1.
The underlying diseases associated with secondary thrombocytopenia were SLE in 16 (34.8%) patients, 11 (68.8%) women and 5 (31.2%) men; infection in 12 (26.1%); thyroid disease in 8 (17.3%), 6 with Graves disease and 2 hypothyroidism; 2 (4.3%) APS; and 2 (4.3%) Evans syndrome. The remaining 6 patients had diagnoses, including HCV, fibromyalgia, psoriatic arthritis, connective tissue disease, papillary thyroid cancer, and one was vaccination-associated. H. pylori testing was performed in 12 patients and 6 (50%) were positive.
Of the 46 adults, 10 (21.7%) were treated exclusively with steroids, 15 (32.6%) received rituximab at low doses (100 mg/m2/week/4 weeks) plus steroids, 8 (17.4%) danazol plus steroids, 1 (2.2%) eltrombopag plus steroids, 2 (4.3%) eltrombopag plus steroids plus low doses of rituximab, 2 (4.3%) IVIG plus steroids, 4 (8.7%) rituximab plus danazol plus steroids, and 4 (8.7%) were under clinical observation. Response was achieved in 95.65% of the patients. CR was documented in 20 (43.48%) and R in 24 (52.17%); there were 2 (4.35%) NR diagnosed with secondary ITP (SLE-associated) and (thyroid disease-associated). The median time to maximum response was 6 (1-45) days. Responses to each treatment are summarized in Table 3. Adverse effects of treatment were observed in 3 adults, including hyperglycemia, acne, and weight gain.
Table 3 Initial response in 46 adults with secondary immune thrombocytopenia at Dr. José Eleuterio González University Hospital in Mexico
| Treatment | Complete response, n (%) | Response, n (%) | No-response, n (%) |
|---|---|---|---|
| Steroids alone | 5 (50) | 4 (40) | 1 (10) |
| Rituximab plus HDD | 5 (33.3) | 9 (60) | 1 (6.6) |
| Danazol plus steroids | 2 (25) | 6 (75) | |
| Eltrombopag plus HDD | 1 (100) | | |
| Eltrombopag plus rituximab + HDD | 1 (50) | 1 (50) | |
| Rituximab plus danazol + HDD | 2 (50) | 2 (50) | |
| IVIG plus steroids | 2 (100) | | |
| Clinical Observation | 2 (50) | 2 (50) | |
HDD: High dose dexamethasone; IVIG: intravenous immunoglobulin G.
The cohort was further divided into 3 groups according to the associated disease; 16 (34.8%) SLE, 12 (26%) infection, and 18 (39.1%) miscellaneous causes mentioned above. Secondary ITP (SLE-associated) patients and those in the miscellaneous group presented a drop in the platelet count at 4 months of follow-up with a median decrement of 96 and 100 × 109/L, respectively. Nonetheless, the median platelet count remained higher than 100 × 109/L. The platelet count course is shown in figure 1.
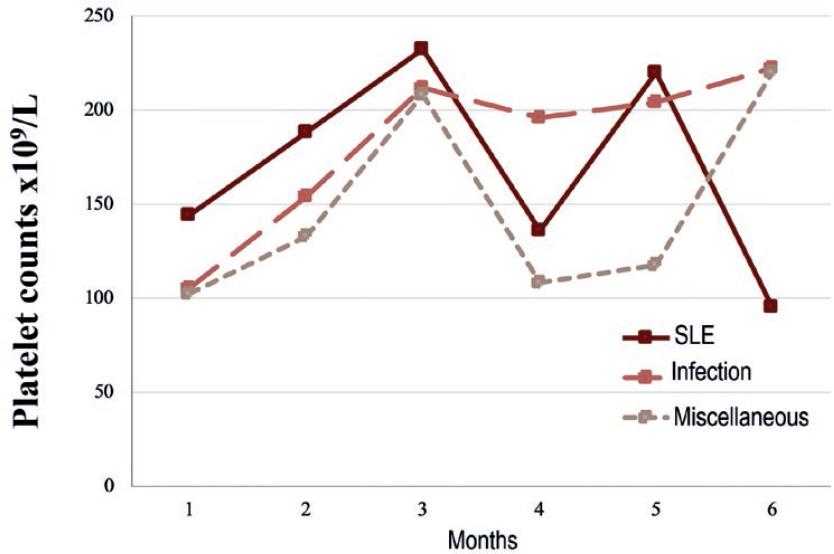
Figure 1 Median platelet count during the first 6 months of follow-up in 46 adults with secondary immune thrombocytopenia (ITP) associated with systemic lupus erythematosus (SLE), infection, or miscellaneous diseases. SLE-associated ITP had the worse course.
Fifteen (34.1%) of the 44 patients that reached a response relapsed at a median of 2 (0.2-70) months; 6 (40%) were treated again with steroids, 8 (53.3%) with rituximab plus high-dose dexamethasone; and 1 (9.1%) had a splenectomy. Fourteen (93.3%) achieved a second response, except for 1 (6.7%) patient with an underlying infectious disease receiving steroids only.
At last follow-up, 4 (8.69%) adult patients had persistent disease, and 14 (30.43%) progressed to a chronic course, 8 (57%) women and 6 (43%) men. In those who followed a chronic course, 5 (35.7%) had SLE, 4 (28.5%) thyroid disease, 2 (14.3%) Evans syndrome, 2 (14.3%) infection, and 1 (7.1%) APS, displayed in figure S1. The median follow-up for adults was 26 (range: 1-157) months. The clinical course according to a secondary cause is shown in Table S2. No significant difference was found among the three groups.
By logistic regression analysis, no factor was found to be statistically significant for chronicity in adults with secondary ITP, Table S3.
DISCUSSION
ITP is a relatively common disease in the geographic region of the study, being the second and third most frequent hematological diagnosis in children and adults, respectively11. Secondary ITP prevalence varies greatly worldwide, with an estimated 20% of all ITP cases in the United States compared to 32% in the present report10. Studies dealing with demographic characteristics, clinical findings, treatment alternatives, and long-term outcomes in secondary ITP are scarce, with a predominance of information on its primary form. Findings in our study cohort contribute to define these features.
We gathered information on 83 patients of all ages over a decade. Secondary ITP was slightly more common in adults and females, as reported in previous studies12. The median platelet count at diagnosis was 8.14 and 13.1 × 109/L in children and adults, respectively, considerably lower than 18.1 × 109/L and 25.4 × 109/L reported in primary ITP13. It is known that most children do not have serious bleeding problems despite low platelet counts, as confirmed in our study where children presented lower platelet counts and a median WHO bleeding scale of 1 compared to 2 in the adult group5. These findings support theories of a higher platelet threshold for bleeding in children, probably due to enhanced platelet function14.
The CR to first-line treatment was 93.97%, higher than 70-90% reported in primary ITP5,15. The median time to response was 6-7 days, considerably faster than the 15 days reported in a study where only eltrombopag was used12. In the present study, eltrombopag was successfully administrated combined with steroids or with steroids plus low dose rituximab in three adult patients; all achieved an initial response. These treatment regimens have been previously studied as first-line treatment for primary ITP with encouraging results, but further evaluation is needed regarding its role in secondary ITP16,17.
Chronic ITP ensued in 30% adults and 19% children, lower than 50-70% and 20-25% reported in primary ITP, respectively7,18,19. To the best of our knowledge, there are no previous studies regarding progression to chronic disease in secondary ITP; some authors suggest its course is expected to be more complicated than in the primary disease, our study documents the contrary3,4.
Risk factors for chronicity in children were age >10 years, as previously reported in primary ITP, and platelet counts ≥20 × 109/L at diagnosis19. Paradoxically, an increased platelet count at diagnosis as a risk factor for chronicity could be associated to an epitope spreading mechanism upregulated by the underlying disease. This could relate to the overall lower incidence of chronic ITP in our study with the lower platelet counts presented in our patients compared with primary ITP studies in other populations13,20.
Depending on the underlying disease, a great diversity of pathways lead to immune dysregulation. These pathways can be divided into central (bone marrow) defects, as seen in autoimmune diseases, or peripheral defects associated with chronic underlying conditions such as infections10. It is thought that peripheral defects have a better response rate to treatment and our study appears to support this concept (83.7% in SLE vs. 100% in infections). However, this difference did not reach statistical significance. In addition, chronic ITP was more frequent in SLE-associated and thyroid disease-associated ITP than in the infections group, supporting the rationale of a more severe immune dysregulation associated with autoimmune diseases than a transitory peripheral defect in acute and chronic infections10.
ITP appears as a secondary complication in 10-40% of SLE patients, while 25% of secondary ITP cases are associated with SLE10,21,22. This is comparable to 24% observed in our study and expectedly lower than the estimated 61.9% when considering all forms of rheumatic diseases reported in Mexico23. Thyroid disease is associated with 5% of secondary ITP cases10, whereas we found a prevalence of 17.4%. Evans syndrome is an uncommon disease reported in 0.7-2% of secondary ITP cases; in our report, it accounted for 3.6%, closer to the 4% found in a Pakistani population10,22,24.
Infection-associated ITP was found in 53% (44/83) of our entire cohort, similar to 52.4% estimated in an all-age electronic survey in Mexico23. Children were more prone to infection-associated ITP; 32 (86.4%) referred to a previous acute infection and 75% of them achieved complete remission, comparable with the prevalence in children of 66% with a remission rate of 80% reported in other series4,10.
H. pylori infection prevalence varies worldwide, with ranges from 1% in the United States to 60% in Japan and Italy4,10. In this study, H. pylori test was performed when the primary disorder remained unclear; therefore, only 12 patients were tested and 50% (6/12) presented a positive result, similar to 60% (14/23) found in a Mexican population25, and higher than 31% in an international study and 5% in the United States10,13. In addition, SLE patients have been reported to have a 39% prevalence of H. pylori infection26. In this study, only three SLE patients were tested with two positive results (66.6%). The benefits of routinely testing for H. pylori and its eradication need to be further studied.
Only one case (2%) was vaccination-associated, it presented on a 32-year-old woman with a recent hepatitis B and influenza vaccination in the previous 5 days. Vaccination has been reported to account for 5% of secondary ITP cases4,10. There are few case reports of influenza vaccination associated with ITP10,27,28, and it is unknown whether simultaneous vaccination leads to an increased risk for ITP.
Adult patients from the SLE and miscellaneous groups presented a drop in median platelet counts at four months of follow-up, as opposed to the infections group, which had a more consistent platelet response. Interestingly, this equals the median time to relapse of 4 months seen in the SLE-associated group of the present study. Another report found a less fluctuating median platelet count through time; however, the platelet range in that study was notoriously wider around the 4th month of follow-up12. Therefore, stricter control during the first 4 months of treatment could be warranted.
Secondary ITP associated with autoimmune diseases was notoriously more prone to follow a chronic course than when associated with infections, probably due to the ongoing autoimmune process; this supports the theory of different pathways triggering a similar clinical presentation. Further studies are needed to deepen the understanding of these mechanisms, which could lead to improved targeted treatment according to the primary disease.

