











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCTION
Human papillomavirus (HPV) infection represents one of the most prevalent sexually transmitted infections, particularly in the sexually active population, but only 10% of cases will develop a persistent infection1. HPV infection persistence is recognized as a requirement for the development of invasive cancer2. This implies that the majority of infected individuals have effective defense mechanisms to eliminate the initial HPV infection.
High-risk HPV E6 and E7 oncoproteins are known to contribute to cervical carcinogenesis by inactivating tumor cell suppression proteins, mainly p53 and retinoblastoma protein (pRb), therefore, prolonging the cell cycle with apoptosis suppression and predisposing cells to neoplastic transformation. In addition, viral oncoproteins have been reported to be able to interfere with components of the immune response1. This review describes the different mechanisms used by the HPV-transformed cells aimed to evade immune recognition and elimination.
A review of the literature was performed employing the PICO acronym to detect articles encompassing immune response, HPV, and cervical cancer (CC). Key words used in the PubMed database included “CC,” cancer,” “HPV,” “immune evasion,” “T cell activation,” “CD4,” “T-cell receptor (TCR),” “HPV E6,” “HPV E7,” “HPV infection,” “Cytotoxic T Lymphocyte-Associated Protein 4 (CTLA-4),” “programmed death protein-1 (PD-1),” “programmed death ligand 1 (PD-L1),” “therapeutic target,” and “immunotherapy.” Manuscripts focusing in Mexican population were detected and others from different regions were also included, articles published in English language over the past 15 years were included to ensure scientific validity.
Articles were revised by all authors, and the information processed according to the GRADE system. Evidence was classified and then recommendations were posed according to the strength of the evidence.
CELLULAR IMMUNE RESPONSE AGAINST HPV-ASSOCIATED CERVICAL CANCER
HPV reaches the epithelial basal layer through microlesions, where physical barriers play an important role in preventing HPV infection in basal cells (Fig. 1). The presence of mucoproteins, an acidic pH, as well as certain human defensins such as HD5 prevents the virus from entering the keratinocyte3. However, oftentimes, HPV can evade these mechanisms and infect its target cell. Once infection is established, the first line of defense against HPV is mediated by the innate immune response. To initiate an effective defense against infected cells, it is important to trigger a rapid inflammatory response4. In the early response, cell recruitment takes place and includes dendritic cells (DC), Langerhans cells (LC), natural killer (NK) cells, and NK T cells (NKT) at the site of infection. All these cells are also involved in promoting an immune response against infection, and most can foster a cytokine-mediated pro-inflammatory process. Among these, type I interferons (IFNs) are involved in the activation of adaptive immune response cells by regulating cytotoxic T cell differentiation, DC maturation, and NK cell activation5. In patients with CC, the HPV E7 oncoprotein has been shown to inhibit the expression of genes induced by IFN regulatory factors. This blocks IFN-alpha activity, which prevents the release of inflammatory cytokines at the site of infection as well as DC maturation and consequently, inhibits the T lymphocyte-mediated cytotoxic response6. In addition, in response to the presence of HPV16 virus-like particles in CC, plasmacytoid DCs have been observed to secrete several cytokines such as IFN-alpha, interleukin 6, or tumor necrosis factor-alpha7.
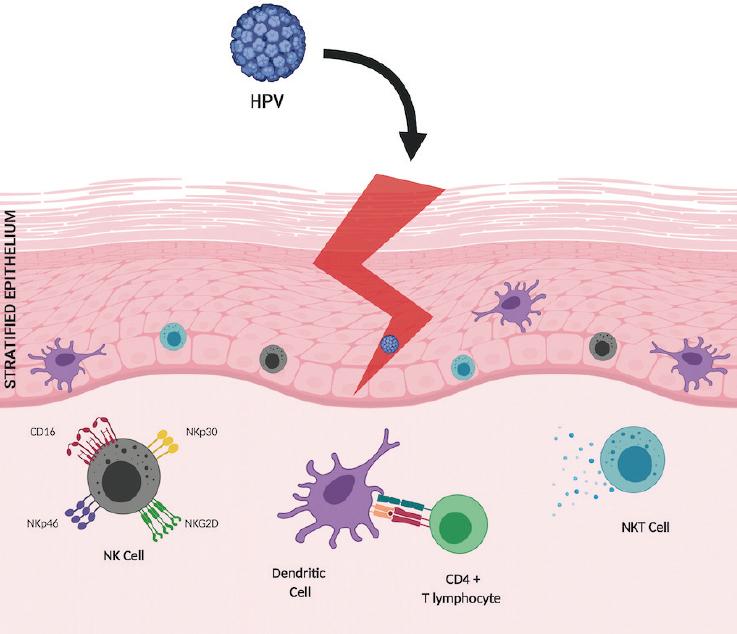
Figure 1 Human papillomavirus (HPV) reaches the epithelial basal layer through microabrasions. Once the infection is established, the first line of defense against HPV is the innate immune response, where the recruitment of Langerhans cells, dendritic cells, NK cells with their varied repertoire of activation receptors, and natural killer T cells (a type of T lymphocyte aimed at innate immunity) takes place, with rapid production of antiviral cytokines such as interferon-γ. Subsequently, a highly specific response to eliminate the infection will include CD4+ T lymphocytes polarized to a T-helper lymphocyte 1 phenotype and induction of a cytotoxic response mediated by cytolytic CD8+ T lymphocytes.
Given that the HPV life cycle is strictly intraepithelial, the immune response of host cells, basal keratinocytes, is necessary to promote HPV elimination. Basal keratinocytes are considered immune system sentinels by acting as non-professional antigen-presenting cells (APCs) and by inducing the expression of T-helper lymphocyte 1 and Th2-type cytokines, as well as a CD8+ T lymphocyte-mediated cytotoxic response. In the female genital tract, keratinocytes express several Toll-like receptors (TLRs), which are capable of recognizing pathogen-associated molecular patterns. The presence of non-methylated viral DNA activates these receptors, triggering the innate and adaptive immune responses, which, in turn, promote cytokine production and create a pro-inflammatory environment with the ultimate goal of eliminating the infection4,8.
When removing infected cells through the cytotoxicity mechanism, major histocompatibility complex (MHC) Class I (MHC-I) molecules intervene and are recognized by NK cells and cytotoxic T lymphocytes. NK cells are able to recognize and eliminate HPV-infected cells, and therefore, functional deficiencies in NK cells have been associated with increased HPV infections and a higher incidence of related cancer9. Cells that exhibit normal expression of MHC-I molecules are protected from the activity of NK cells; however, HPV-infected cells show a decrease in MHC-I expression induced by viral oncoproteins, which leads to their elimination by NK cells. In addition, once viral proteins are processed by the immunoproteasome/endoplasmic reticulum, they form stable complexes with the MHC-I heavy chain and beta-2-microglobulin accessory protein, which migrate to the cell surface and induce a specific cytotoxic response by CD8+ T cells and infected cell lysis10.
In some tumors, it is common to observe changes in MHC-I and II expression, and especially a decrease in the expression of MHC-I molecules. In fact, HPV6 E6 oncoprotein reduces MHC-I protein levels by inhibiting the expression of MHC-I genes. Apparently, HPV16 E6 can modulate MHC-I expression, unlike HPV18, which is unable to do it, and thus promote the suppression of the adaptive immune response11. In addition, the HPV E5 oncoprotein also regulates the levels of MHC-I complexes by interfering with their positioning on the cell surface, thereby preventing the recognition of infected cells12.
In summary, HPV infection promotes a rapid and short-lasting inflammatory response, as well as the recruitment of cells involved in immunity. The presence of the virus induces a specific cytotoxic response mediated by MHC-I that leads to infected cell lysis. However, in some susceptible women, the virus has mechanisms to evade the immune response by decreasing MHC-I expression or blocking IFN activity, which generates an immunosuppressive microenvironment, in which immunosuppressive cytokines such as IL-10 and transforming growth factor beta (TGF-beta) participate13.
HPV manages to evade the immune response by preventing and minimizing its exposure to the immune system. HPV replication and release do not cause cell death, given that differentiated keratinocytes are already programmed to die; furthermore, HPV takes advantage of the keratinocyte differentiation program to complete its viral cycle in the superficial layers where the assembly of new virions takes place and is released by the action of the viral protein E4 during the natural desquamation process14, so a danger signal activating the immune system is not generated. On the other hand, HPV minimizes the expression of capsid proteins and delays their expression in the differentiated epithelium to avoid or delay their detection by LCs, which allows the establishment of persistent or latent infection in host cells. In patients with CC, dead cells released during normal turnover of the cervical epithelium are phagocytosed by LCs, which release immunosuppressive cytokines such as TGF-beta, IL-10, and IL-13. Furthermore, through the effect of these cytokines, HPV infection prevents the activation and maturation of DCs and inhibits the triggering of the T lymphocyte positive cytotoxic response15-18.
Another mechanism through which recognition is prevented, in infected or transformed cells, is the absence of costimulatory molecules and T lymphocyte activation receptors. Costimulatory molecules identify and eliminate transformed cells. Several studies have established the ability of HPV to regulate the type and amount of these molecules in HPV-positive cells. Tummers et al. (2014) determined the importance of CD40 activation during inflammatory and antitumor responses, reporting that after 48 h of ligation, a phenotype that is necessary for the development and maintenance of the adaptive immune response is promoted, but weakened by the effect of HPV19. CD40 expression is promoted by the transcriptional factor of the AT-Hook AKNA type20, which is a regulation target for HPV. As a result of this dysregulation, CD40 and IL-8 are decreased21. In addition, HPV is capable of modifying cytokine levels as a mechanism of evading the immune system, thus inhibiting the pro-inflammatory response in keratinocytes. High-risk HPV E6 and E7 oncoproteins inhibit TLR9 expression, which limits their ability to induce the expression of pro-inflammatory genes that are essential for an immune response against viral infection22.
NK cells are known to be important effectors of early immune surveillance of tumors. The activity of these cells is finely regulated by the balance between inhibition receptors, whose main ligands are MHC-I molecules, and activation receptors, which partially recognize ligands induced after cellular stress23. In the particular case of CC, the loss of MHC-I caused by HPV oncoproteins would leave the tumor cell susceptible to the cytotoxic effect of NK cells. However, NK cells in these patients have been observed to often be dysfunctional and are, therefore, unable to mount an effective cytotoxic response against the tumor. This dysfunction can be explained by defects in the expression of important activation receptors such as NKp30 and NKp46, which have been observed to be decreased in the peripheral NK cells of patients with CC and, in addition, this decrease is consistent with defects in their cytotoxic activity24. These findings suggest that defects in activation receptors may support CC progression.
The proportion of CD4+ and CD8+ T cells is related to the severity of the lesions in the cervical mucosa. Patients with cervical intraepithelial neoplasia (CIN) in regression or low-risk HPV-induced genital warts have a higher proportion of CD4+ than CD8+ T cells, while in patients with advanced lesions and invasive CC, the proportion of CD8+ is higher25,26. Despite the increase in the number of cells responsible for the elimination of tumor cells, they are anergic and non-functional27. Table 1 summarizes a variety of abnormalities through which CC evades cellular immunity, promoting tumor growth and dissemination.
Table 1 Summary of immune abnormalities in patients with cervical cancer
| Alteration | Consequences | References |
|---|---|---|
| Loss of MHC-I molecule expression in cervical cancer | Defects in the response by cytotoxic CD8+ T lymphocytes | 11,41 |
| Resistance to cytotoxic T lymphocyte-induced lytic activity | ||
| Altered cytotoxic CD8+ T lymphocytes in cervical neoplasm | CD4/CD8 ratio is altered in favor of CD8 T lymphocytes Decrease in IFN-γ and IL-5 | 25 |
| Decreased TCR/CD3 complex zee chain in T lymphocytes | Defects in the signals generated through TCR/CD3, which affect the proliferation and production of cytokines | 27 |
| Decreased E-cadherin in cervical cancer cells | Defects in the retention of Langerhans cells in the epidermis and subsequent immune masking | 42 |
| Death by apoptosis of CD4+ and CD8+ effector T lymphocytes | Apoptosis induction and T lymphocyte proliferation inhibition through the production of soluble factors | 43,44 |
| Increase of immunosuppressive cytokines in the cervix | Suppression of specific immune responses in the tumor microenvironment by IL-10 and TGF-beta | 17,18 |
| Maintenance of viral persistence and transformed cells | ||
| Increased Treg lymphocyte recruitment in the cervix | Destruction of tumor immune surveillance by counterattacking tumor infiltrating T lymphocytes (TIL) | 45,46 |
| Cytotoxic CD8+ T lymphocyte function inhibition | ||
| Absence of functional CD40 on the surface of tumor cells | Absence of CD40 in APC does not allow T lymphocyte activation, with subsequent decrease in pro-inflammatory cytokines | 21 |
| Loss of CD28 and perforin acquisition in the population of circulating CD4+ T cells in CC | The role of CD4+ CD28null T cells in CC is not clear. | 47 |
| They might act on tumor growth | ||
| CD28 and CTLA-4 dysregulated expression in peripheral T lymphocytes of patients with CC | Altered T lymphocyte function that leads to systemic immunosuppression | 32 |
| Increased expression of PD-L1 in tissues of patients with CC and of PD-1 in stromal mononuclear cells (identified as TIL) | Overexpression of the PD-1/PD-L1 axis, one of the main pathways of immune control in T lymphocytes, leads to a potent inhibition of antitumor immune responses | 48,49 |
HPV: human papillomavirus; MHC: major histocompatibility complex; CTLA-4: cytotoxic T lymphocyte-associated protein 4; PD-1: programmed death protein-1; PD-L1: programmed death ligand 1; TCR: T lymphocyte receptor.
IMMUNE CHECKPOINTS: CTLA-4, PD-1/PD-L1, AS IMMUNE MOLECULAR TARGETS IN CERVICAL CANCER
The immune response against virus-infected cells and against tumor cells is regulated by the interaction between APCs and T lymphocytes (Fig. 2A). APCs express on their membrane a series of costimulatory and corepressor molecules, which bind to the receptors present on the antigen presentation-activated T-cell membrane. These molecules are known as immune checkpoints and, as a whole, they are responsible for maintaining homeostasis after an immune response28.
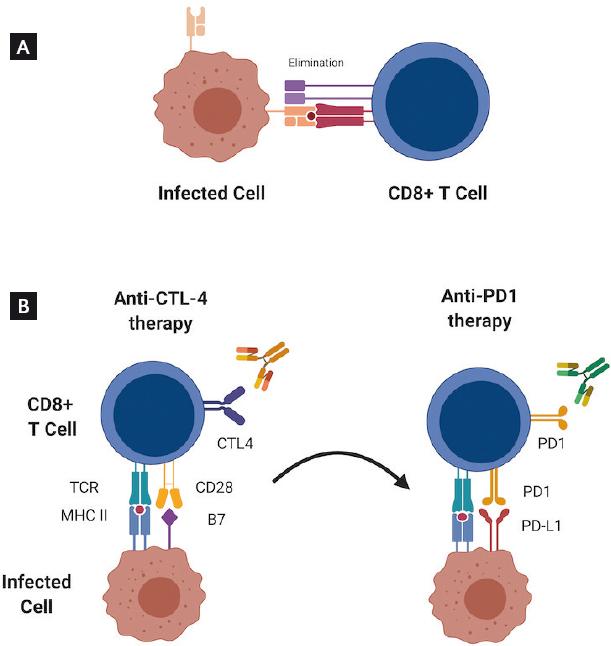
Figure 2 Elimination of infected cells and tumor cells. A. Induction of a cytotoxic response mediated by cytolytic CD8+ T cells against infected cells. B. Under certain circumstances, the use of anti-cytotoxic T lymphocyte-associated protein 4 antibodies might promote the expression of programmed death protein-1 receptors, a strategy that could be exploited as a therapeutic target in the treatment of cervical cancer.
CTLA-4 AND CERVICAL CANCER
The activation of checkpoints in T lymphocytes is a sequential phenomenon, and their function is not redundant29, so after antigen recognition by the TCR, binding of the CD28 receptor to its ligands (CD80 and CD86) occurs in APCs. This induces positive signals for T lymphocyte activation, but also favors the translocation of the CTLA-4 inhibitor receptor to the lymphocyte membrane. CTLA-4 is homologous to the costimulatory protein CD28 which it competes with, but its affinity is approximately 20 times higher for the CD80/CD86 ligands30. By binding to its ligands, CTLA-4 activation induces its association with phosphatases that inactivate molecules such as the TCR CD3-zeta chain28 and Akt31, thus inhibiting the signaling cascades that promote cytotoxic T lymphocyte activity.
The clear association between cellular immune activity and the natural response to HPV infection and its associated lesions suggest that during the development of CC, there are cell response failures that limit the recognition and elimination of tumor cells. An analysis by Kosmaczewska et al. showed that an abnormally high proportion of circulating CD4+ T cells from patients with CC express CTLA-4, while a significant decrease in CD28 expression is observed in CD8+ T cells32. These observations appear to indicate that the T-cell-mediated response against tumor cells could be inhibited by the presence of CTLA-4 and suggest that patients with CC could benefit from immunotherapy directed at blocking CTLA-4 (Fig. 2B).
As a result, a Phase I-II clinical trial was conducted to explore the possible anti-tumor effect of ipilimumab in patients with HPV-associated metastatic or recurrent CC33. Ipilimumab is a humanized monoclonal antibody that blocks the membrane CTLA-4 molecule with high affinity and was approved by the FDA in 2010 for the management of different types of cancer34. The reported study is a multicenter protocol that recruited 42 patients with HPV-positive CC and resistant to cisplatin. The efficacy of ipilimumab as monotherapy was tested and the results showed that CTLA-4 blockade, targeted therapy did not increase patient overall survival or the progression-free interval, and therefore, patients with CC were considered not to be benefited by this type of immunotherapy. However, an analysis of the activation status and the expression of markers in circulating lymphocytes showed that treatment with ipilimumab induced the expression of the inhibitory molecule PD-133, which opens a new perspective and the possibility of immunotherapy in CC, using antibodies targeting PD-1.
PD-1/PD-L1 AND CERVICAL CANCER
Another immune checkpoint that has drawn attention in recent years is undoubtedly the PD-1 molecule, initially described in the T-cell population; however, PD-1 is also known to be expressed in B cells, mainly NK and DC cells. Two PD-1 ligands, PD-L1 and PD-L2, both members of the B7 family, are known. PD-L1 can be induced by various cytokines, such as IFN-alpha, and its expression encompasses a wide variety of cells both hematopoietic (including APC, such as DC), and non-hematopoietic in nature (e.g., tumor cells and tumor-associated fibroblasts), while the expression of PD-L2, which is induced by IL-4 and IFN, is restricted to DCs, macrophages, mast cells, and some B cells30.
The interaction between PD-L1 in APCs and its PD-1 counterpart in activated T lymphocytes leads to the inhibition of the latter cells, which results in apoptosis induction or leads to a state of anergy, which inhibits the production of cytokines and cytolytic function35. In addition, PD-1 expression enhances the conversion of immature CD4+ T cells into regulatory T cells (Tregs), causing immune response attenuation. The PD-1/PD-L1 axis is known to represent a novel therapeutic target in different tumors; however, the clinical significance of PD-L1 expression in CC has not been fully elucidated. For example, PD-L1 overexpression has been reported in a variety of malignant tumors, causing cytotoxic CD8+ T cells present in the tumor microenvironment to shut down their lytic function, and thus, it becomes a protective mechanism against cell death.
At present, there are several studies that have reported the expression of PD-L1 in CC samples. For example, HPV16 E7 expression has been associated with PD-L1 expression in CC cells35. A recent study in patients with CC showed PD-L1 expression in 34% of samples; when the samples were subcategorized, a larger number of positive samples were observed in squamous cell carcinoma (38%), in comparison with adenosquamous carcinoma and endocervical adenocarcinoma (29% and 17%, respectively). Notably, PD-L1 was not found in normal cervical samples or in benign cervical lesions36,37. Another study showed PD-L1 positivity in > 5% of tumor cells and in 54% of squamous-cell carcinomas versus 14% for adenocarcinomas (p < 0.001). Interestingly, in that same study, positivity for PD-L1 was also found in tumor infiltrating immune cells, as well as in stromal immune cells, most of them identified as tumor-associated macrophages38. Finally, another study showed that PD-L1 expression is associated with efficient HPV infection and that it is also significantly overexpressed in both CC cells and surrounding inflammatory cells in comparison with other gynecological tumors39.
Overall, the discovery of this important negative regulatory pathway in the biology of T cells has opened new therapeutic options, such as immunotherapy with the use of antibodies targeted against PD-1 or PD-L1, designed to block the interaction between the receptor and its ligands, which has shown established clinical benefit in a variety of tumors, including CC; these are currently being investigated in Phase I/II clinical trials. Therefore, directing therapies against multiple immune pathways, in particular, with antagonists of the PD-1/PD-L1 pathway (Fig. 2B), might overcome tumor resistance to immune effector mechanisms, especially in advanced or recurrent tumors (Table 2).
Table 2 Immunotherapy (alone or combined) against cervical cancer: current clinical trials based on the use of antibodies targeting immune checkpoints or their ligands
| Registry number | Treatment | Type of antibody | Target | Phase | Type of tumor | Reference(s) |
|---|---|---|---|---|---|---|
| NCT01711515 | Ipilimumab | Human monoclonal antibody (IgG1) | CTLA-4 | I | Stage IB2-IIB or IIIB-IVA cervical cancer | 50 |
| NCT02635360 NCT02628067 | Pembrolizumab | Humanized monoclonal antibody (IgG4) | PD-1 | II | Advanced cervical cancer | 50,51 |
| NCT03298893 | Nivolumab | Human monoclonal antibody (IgG4) | PD-1 | I | Locally advanced cervical cancer | 52,53 |
| NCT02921269 | Atezolizumab | Humanized monoclonal antibody (IgG1) | PD-L1 | II | Recurrent, persistent or metastatic cervical cancer | 52 |
CTLA-4: cytotoxic T lymphocyte-associated protein 4; PD-1: programmed death protein-1; PD-L1: programmed death ligand 1.
OTHER IMMUNE CHECKPOINTS AS POSSIBLE THERAPEUTIC TARGETS FOR CERVICAL CANCER
As previously mentioned, NK cell-mediated cytotoxic activity is pivotal to the immune response against HPV-infected cells and CC cells. Since abnormalities in NK cell function have been observed during the development of CC, it is reasonable to consider that this type of cancer could be a candidate for treatment with new immunotherapies targeting molecules which inhibit NK lymphocyte function as, for example, some killer cell immunoglobulin-like receptors (KIRs) which are overexpressed in CC and that is currently under development40.
CONSIDERATIONS
According to our current knowledge on immune responses in CC and to the critical elements that are compromised during the development of HPV-associated tumors, CC is considered to be a good candidate for treatment with immunotherapeutic strategies. At present, there are three commercially available immunotherapeutic options that use antagonist monoclonal antibodies: (1) against CTLA-4, (2) against PD-1, and (3) against PD-L1. In the case of CC, CTLA-4 expression has been reported in a high proportion of analyzed samples; however, this treatment’s efficacy proved to be of little benefit to patients, but the use of CTLA-4-targeted therapy induced PD-1expression. This is relevant, because there are data indicating that not all patients with CC express PD-1, and thus would not be candidates for therapy targeting this molecule and/or its receptor.
Overall, this opens the possibility of establishing sequential treatment strategies using existing commercial antibodies. Furthermore, knowledge of the presence of other altered immune checkpoints in CC, such as KIRs, suggests that this type of tumor should be considered as a model to prospectively study the efficacy of therapies that are currently under development and that use this checkpoint as a target.
CONCLUSIONS
The immune response plays a preponderant role in the elimination of CC cells. Failures in critical points of the cellular immune response foster the development of tumors. Given the information described, CC is a candidate for immunotherapy treatment.
Recommendations
Altered CTLA-4 expression has been observed in CC. Therapy aimed at blocking CTLA-4 has not been successful; however, CTLA-4 blockade induced PD-1 expression, which suggests that a sequential treatment strategy might favor the response of these patients when treated with combined therapies, targeting both CTLA-4 and PD-1. Quality of evidence: (GRADE) moderate. Strength of recommendation: strong in favor of its use.
The degree of response with the use of immunotherapy will largely depend on PD-1 and PDL-1 targets’ expression. Therefore, it is necessary to determine the degree of expression of these molecules to ensure therapeutic success. Quality of evidence: (GRADE) high. Strength of recommendation: strong in favor of its use.
It is advisable to include CC as a candidate for new therapies. KIRs provide a strong strategy in the treatment of cancer. Therefore, studies with anti-KIR antibodies might prove to be a promising strategy for the treatment of CC. Quality of evidence: (GRADE) low. Strength of recommendation: weak in favor of its use.
The study of possible therapeutic combinations for CC represents an opportunity to provide new treatment regimens to decrease adverse effects and improve treatment responses. Quality of evidence: (GRADE) low. Strength of recommendation: weak in favor of its use.
It is necessary to delve into the molecular mechanisms involved in cellular immune response control in cervical tumors to detect possible novel therapeutic targets. Quality of evidence: (GRADE) high. Strength of recommendation: strong in favor of its use.
The implementation of efficacious methods in the production and purification of immunotherapeutic agents will potentially provide tools for a rapid and successful implementation of immunotherapy. Quality of evidence: (GRADE) low. Strength of recommendation: weak in favor of its use.

