











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkBACKGROUND
The pharmaceutical industry has become the primary source of studies able to provide the highest quality data to build evidence-based guidelines1. Due to their high cost, the same trial will be used to obtain regulatory approvals of the study drug, and the cost-effectiveness information needed for authorization of reimbursement by health agencies. As a result, the complexity of a study is extraordinary in many ways. Pharma investigators have developed highly structured networks in which every process should be observed and verified. Participation in industry-sponsored studies is beneficial to learn and implement good clinical practices (GCPs). However, in some cases, the study design is pushed to the limit causing ethical dilemmas. Research Ethics Committees (RECs) are the first line of protection to prevent unacceptable risks for study participants, researchers, and institutions. A balanced distribution of potential benefits should be guaranteed2. The objective of this paper is to review the most common ethical dilemmas found in Pharma-sponsored trials and to propose possible solutions.
THE REFERENCE GROUP DOES NOT RECEIVE THE STANDARD OF CARE. THE ETHICAL DILEMMAS TO BE DISCUSSED ARE
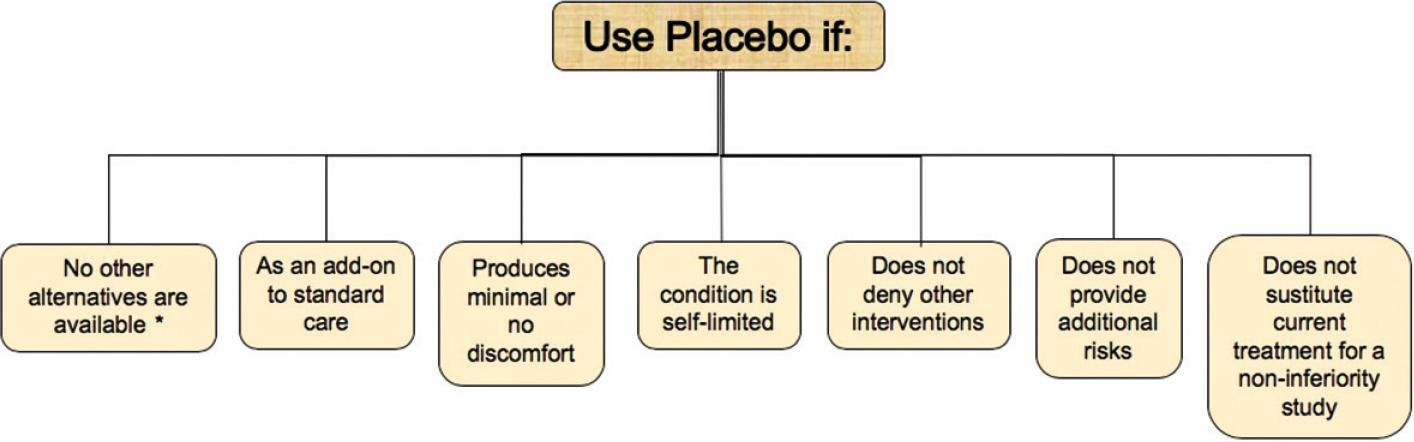
The primary interest of clinical research is to gain knowledge that may improve the existing options for treatment of the problem in question; at the same time, safety and quality of life of the participant should be preserved3. For many years, the effects of new therapies were compared against a placebo. Side effects are more likely to be detected when one arm of the study is assigned to receive a placebo. Furthermore, the sample size required to obtain statistically significant differences is remarkably smaller in placebo-controlled trials. However, scenarios in which this practice is now acceptable to have been limited by the introduction of effective and safe therapies for a large number of diseases4. The use of placebo is considered ethical for conditions in which no diagnostic, preventive, or therapeutic tool is available (Fig. 1). Add-on treatments or reasons that preclude the extrapolation of the evidence to a particular group could be considered as potential indications for the use of a placebo in conditions in which an optimal therapy does not exist5. Other likely indications are self-limited diseases or conditions that cause minimal discomfort, especially if the response to therapy or placebo is highly variable. Finally, some authors6 consider it acceptable when all three of the following conditions are present: compelling methodological reasons, participants are not deprived of interventions they would otherwise receive, and research intended to develop interventions that will benefit the host population. The Declaration of Helsinki states that Where for compelling and scientifically sound methodological reasons, the use of placebo is necessary to determine the efficacy or safety of an intervention and the patients who receive placebo or no treatment will not be subject to any risk of serious or irreversible harm7. Despite the above, the application of these standards could be highly subjective. Examples of areas in which the use of placebo is a matter of controversy are the development of vaccines or psychiatric drugs. The World Health Organization has published position papers that may help RECs make a fair evaluation in these projects8,9. Less clear indications are the impossibility of having access to therapy in low-income countries or health systems in which the therapy is not available. As a result, the Pharma industry has selected low- and middle-income countries or public hospitals to increase the likelihood of acceptance by the use of placebo.
Tools to overcome the problem
RECs should always ask the investigators and sponsors to compare the intervention against the best available option. Participants should receive a benefit from their participation; it may come from the use of the experimental drug or the overall treatment of the condition. A non-inferiority study is an ethical duty for the majority of the placebo-controlled trials. RECs should always ask themselves if the study design was acceptable in high-income countries. If the answer is negative, the benefit that the participants will receive should be established.
Another common ethical misconduct is the switch of effective therapy for a new one that is being developed. When the objective of the switch is to avoid an adverse effect or to simplify an established treatment, this intervention is justified. However, it is ethically questionable to conduct a switch study with the sole purpose of gaining evidence to introduce a new drug to the market without a clear benefit over the existing one. In the same context of switch studies, using noninferiority as the primary objective with lower confidence intervals limits of −10% has been a practice long used in the development of new drugs10. Due to ethical questions about this practice, the new recommendations by Food and Drug Administration (FDA) changed recently to use a more rigorous −4% lower boundary for this type of studies11.
ECONOMIC INTERESTS DRIVE THE AIMS OF THE STUDY
Phase III and IV studies are among the most common sponsored trials conducted in developing countries (Table 3). The purpose of phase III studies is to compare the efficacy and side effects of the experimental therapy against the current standard of therapy for a well-defined condition. Phase IV focuses on efficacy, effectiveness, real-life use, and safety of a drug or treatment already available in the market. Their corresponding study designs are highly variable. Examples of that are: add-on (one or more experimental therapies administered to participants receiving the first-line therapy), crossover, non-inferiority, superiority, and group sequential (a treatment arm stopped if there is evidence of superiority or unacceptable risk compared to the established therapy), and parallel and randomized withdrawal trials (cases with a positive response are randomized to the intervention or placebo). These studies are highly relevant because the results will determine if the drug is introduced in the market and will be the evidence to estimate the cost-effectiveness of the intervention, needed for approval of reimbursement by health agencies. However, ethical misconduct is not rare and could be prevented by the RECs. Sometimes, the merit of the report may be limited to demonstrating a lower cost, a higher adherence rate, or greater satisfaction with the intervention. Participants may not receive a direct benefit; in contrast, they may be exposed to unnecessary risks (by stopping established therapies, by changing the dose regimen, or by having exams or procedures unnecessary for the treatment of the medical condition).
Table 1 Online training options for patient protection programs
| Online training | Web site |
|---|---|
| The Office of Research Integrity | http://ori.dhhs.gov/education/ |
| The National Institutes of Health | http://grants.nih.gov/training/responsibleconduct.htm |
| The Office for Human Research Protections of the US Department of Health and Human Services | http://www.hhs.gov/ohrp/education/ |
| The Collaborative IRB Training Initiative | www.miami.edu/UMH/CDA/ |
| UMH_Main_Printable/1,2153,6460-300.html | |
| Family Health International Interactive Research Ethics Training Curriculum | www.fhi.org/en/topics/ethics/curriculum/default.htm |
| Consorzio per valutazioni biologiche e farmacologiche | www.gcptraining.cvbf.net |
| CITI program | 7.CITI program: https://about.citiprogram.org/es/ |
| https://about.citiprogram.org/es/ |
Table 2 Objectives of good clinical practice
| Good clinical practice | Actions |
|---|---|
| Protection of study subjects | Review protocol by the REC |
| Review Informed consent process | |
| Comply with the trial protocol | |
| Ensure that: Risk justified by the potential benefits Rights and safety of participants are above the interests of science Privacy of the participants is preserved | |
| Report AEs and SAEs | |
| Obtain certification in good clinical practice of all parties involved | |
| Provide monitoring, audits, and inspections | |
| Quality of the data | Validate information |
| Encourage representativeness and generalizability | |
| Reassure traceability | |
| Perform monitoring, audits, and inspections | |
| Transparency of trial conduct | Obtain contracts between all parties involved |
| Allocate and document responsibilities | |
| Certificate trial conduct | |
| Document generation, gathering, processing, and evaluations of the clinical trial data |
AE: adverse events; SAEs: serious adverse events.
Table 3 Clinical trial phases
| Phase | Primary goal |
|---|---|
| Preclinical | Testing of the drug in non-human subjects |
| Phase 0 | Pharmacokinetics |
| Phase I | Testing of the drug on healthy volunteers for safety; involves testing multiple doses |
| Phase II | Testing of the drug on patients to assess efficacy and side effects |
| Phase III | Testing of the drug on patients to assess efficacy, effectiveness, and safety |
| Phase IV | Postmarketing surveillance. Trials conducted after a drug has been demonstrated to work and has been granted a license. Indicated to increase knowledge on side effects, safety, risks, and benefits in real life. |
The REC should be given references related with results of the previous study phases to analyze whether the drug should be considered in the study phase under review.
Observational studies that intend to evaluate a specific treatment after it was introduced into the market are useful to describe the real-life effectiveness of a drug without the extreme selection of patients which occurs in clinical trials. However, in particular contexts, these studies may be an induction to prescribe the new drug. RECs should request that payments to the investigator or the institution be calculated based on qualitative parameters, instead of on the number of cases enrolled. Participants may be exposed to loss of privacy or side effects that are not covered by the sponsor, or changes in the services covered by their health provider (e.g., by premature discontinuation of the study drug). As a result of their participation, investigators and institutions may have unforeseen conflicts of interest.
Tools to overcome the problem
RECs should be provided with all the evidence generated by the sponsor to measure the risks and benefits of the study. They should have enough information to classify the phase of the study. RECs should request that every study protocol contains a section in which the direct benefit to participants is clearly stated. Potential risks should be identified; preventive/compensatory actions should be foreseen for every hazard. Comparisons between drugs with very similar efficacy and safety profiles (me-too therapies) should be reviewed with great caution to limit the execution of studies without scientific merit, justified only by the economic interests of the sponsor. Review boards should protect patients and researchers from unnecessary risks.
SELECTION OF THE STUDY SAMPLE IS BIASED TO OBTAIN THE BEST POSSIBLE RESULTS
Inclusion criteria applied in the controlled or observational studies may distort the profile of the target population. Cases with the highest probability to have a positive response are included; as a result, the efficacy will be lower when the intervention is applied in real life. Similar selection bias is frequently used to decrease the rate of side effects. This approach limits the external applicability of the conclusions12. Exclusion criteria should be carefully reviewed by the REC to ensure that the potential benefits of the intervention are applied with equality and fairness.
THE CONSENT FORM IS NOT USEFUL FOR THE PROTECTION OF THE STUDY PARTICIPANTS
Many papers have documented the existence of significant gaps in the sponsored clinical studies during the informed consent process13,14. Physicians and their team members are not instructed to obtain informed consent fxulfilling the ethical and legal standards. Deficiencies in the informed consent process could be aggravated by the narrow periods allowed for the inclusion of patients in the Pharma projects. Some sponsors have changed the informed consent form into a legal document that protects them from potential demands. The excessive length and the complex language of the document become significant barriers to fulfill the ethical duties of the informed consent process. Furthermore, the future use of stored biological materials obtained during the study is barely mentioned.
Tools to overcome the problem
RECs should request that all personnel involved in the study is appropriately trained to fulfill the duties assigned by the principal investigator. Most sponsors provide online training courses on GCPs; certificates should be stored in the study file (Table 1). Principal investigators are responsible for ensuring that the informed consent process is well executed. However, it is acceptable to delegate this task to a sub-investigator. Evidence supporting the correct execution of the informed consent process should be recorded in the study notes. In addition, RECs may limit the extension of the informed consent document by asking the principal investigator and the sponsor to avoid unnecessary repetitions. The information should be presented using common words; the text should be comprehensible to a person with elementary education. If biologic materials are stored, the document should clearly state its intended use, the period that the sponsor will store them, and the partners who may have access to the materials or the information. The informed consent document should contain specific sections (or even a complementary document) in which participants state their approval for the use and storage of the biomaterials. Furthermore, the document should describe the actions that the participants should take if they want the removal of their biomaterials from the study. Despite the above, the pharma industry is a significant player in the development of new and improved versions of the informed consent process. Cellphone applications or online interactive versions of the consent form have been developed in which it could be verified that participants understood the study-related procedures, risks, and benefits15. RECs should be aware of the new challenges that could result from their use.
INCENTIVES FOR RESEARCH PARTICIPANTS
Altruism is a principle in which the inclusion of study participants should be built. Pharma studies have applied incentives and commercial strategies to increase the interest of the population in a study or to keep participants engaged. Payments, gifts, free access to medical services, travel vouchers or gift certificates are among the most common incentives. Several deviations may result from the use of incentives. An excessive fee may cause a skewed sample or could be a form of coercion. This issue is highly relevant in low-income countries. Some individuals seek their inclusion in the study as a source of income, exposing themselves to unexpected risks. Participants may hide relevant information to be accepted; they may become full-time study subjects regardless of the risk it may imply. Researchers should identify the motivation of the subject to get involved in the study. On the other hand, researchers may fall in coercion by paying the participants only once the study is completed. Besides, cases and controls may not be paid equally, merely because patients (that is, cases) may have free medical attention. At the same time, principal investigators may have motivations to participate in the study beyond the scientific merit of the project. Principal investigators and the steering committee may have economic or academic benefits due to their involvement in the study.
Tools to overcome the problem
Institutions should have unified medical records in which patients enrolled in research studies could be easily identified, especially those that had several participations. RECs should review all ad messages or documents that will be used to invite subjects to participate in the study. Furthermore, every material that participants could be exposed to should have RECs approval. The size of the payment should cover any study-related expense, but it should not be so large that it becomes the main reason to participate. Some variables could be used to estimate whether the amount of the stipend is ethical. Studies that may not be acceptable in a high-income country, or where subjects decline their participation if they do not receive payment, should be carefully analyzed for the presence of potential unethical conditions3. Payments should be scheduled promptly; they should not be used as a tool to keep the patient enrolled until the end of the study. Patients and healthy subjects should be treated in the same way. All protocols should include a justification for the payment, how the amount was calculated, and how and when payment is made. Payment should be the same for all participants or based on the risk or effort required. Even, lotteries could be used to give a sizeable prize rather than a small gift16. In this case, the value of the prize should be disclosed as part of informed consent. As a final point, we should recognize that, for low-income groups, Pharma studies may be the only option to have access to therapy for patients needing a high-cost medication. Finally, RECs and institutional authorities should analyze the motivation of the researcher to accept the Pharma-sponsored project.
All principal investigators and their teams should declare potential conflicts of interest when the study is submitted for REC approval. Academic misconduct is prevented by the proper selection of the steering committee members. This body should approve any local or international publication or presentation of the results. Steering committee members should be independent, highly recognized researchers, statisticians, and/or ethicists. RECs should not be afraid to request the participation of a Data Safety Monitoring Board (DSMB).
FAIR ACCESS TO POST STUDY BENEFITS
Often there are so many questions about the beginnings and development of a clinical research project in vulnerable populations and so much uncertainty about the outcomes of the research participants that it is difficult and even impractical to visualize the patients future at the end of the project. The ranges of clinical, personal, health coverage, and response to treatment options are as wide as the possible outcomes17. The Council of International Organizations of Medical Sciences established that the sponsor should guarantee all participants access to the study product even after the project is completed, once the efficacy and safety were proven18. This moral consensus, which occurs among members of the scientific community and bioethicists, is close to unanimity in certain diseases such as cancer and infection by the Human Immunodeficiency Virus19,20. However, the regulatory variability of each country leads to many realities. For diseases in which alternative therapies besides the study drug exist, patients may be allocated to safe and effective choices at the end of the study if the quality of treatment is not affected by the substitution. If no treatment choice exists, the sponsor should provide access until the intervention becomes available to the population. Studies evaluating the outcomes that occur in study participants after the end of the pre-specified follow-up period of the research study are almost non-existent21.
Tools to overcome the problem
RECs may request a follow-up survey for trials in which there is no therapeutic alternative to the study drug or in conditions with a high risk of health deterioration if treatment is changed22.
THE STUDY DESIGN MAY CAUSE UNNECESSARY RISKS, UNACCEPTABLE REQUESTS OR MAY AFFECT ACCESS TO CARE
Safety is an unquestionable requirement for all clinical studies. The study design of the Pharma-sponsored studies may induce the appearance of symptoms or deterioration of the control in cases with chronic disorders caused by the running-in period or the use of placebo. Studies may require multiple blood draws or unjustified studies that may produce discomfort to the participants, beyond the demands from the usual care of their condition. All outcomes or symptoms that occur after the inclusion of a participant in the study are considered adverse events. Participants should be protected regardless of the cause; sponsors and the principal investigator should provide proper care even if the event is not related to the intervention under study3. Even more, drug-related side effects could be unexpected and occur after the study is ended. In addition, study participants could be exposed to unforeseen risks. Access of their personal information by unauthorized partners or officers may have an adverse effect for the study participant (e.g., for their health insurance or work status). Furthermore, the primary provider of the health service may not give the usual care due to the patients involvement in the study.
Tools to overcome the problem
RECs are the first line of protection against the exposure of participants to unnecessary risks. Studies should be conducted following GCPs23 (Table 2). The study design should be critically reviewed to prevent the exposure of study subjects to any avoidable risk or discomfort. Pre-specified rescue criteria should be requested. In addition, principal investigators should be allowed to add complementary therapies that keep participants safe without interfering with the study. If no therapies exist, exclusion or elimination criteria should be strict enough to protect participants. RECs should request sponsors to acquire a health insurance plan that cover all side effects. This request may create controversies because it could be challenging to establish whether the event is caused by the disease or by the experimental drug. To prevent conflicts, the RECs should request a list of conditions that the insurance plan will cover regardless of the potential cause. All serious adverse events should be informed to the REC following the GCP standards. RECs should not be afraid to request an extended follow-up protocol if unexpected clinical events occur during the trial. However, in real life, it is improbable that a REC could detect an unforeseen side effect or could integrate the information coming from other centers or trials in which the same intervention is used. Additional research is needed to improve surveillance of studies by federal or international authorities. Beyond a thorough review of the research protocol and the informed consent form, the REC should request a monitoring plan, GCP certification of the study members, and means of communicating with the DSMBs when needed. DMSBs are required when a sponsored multicenter trial has mortality or significant morbidity endpoints or severe adverse events (SAEs) are expected with a new or high-risk therapy, or when very little safety data exist before the study, or if vulnerable populations are involved24. These bodies are composed of independent researchers, statisticians, ethicists, and community representatives. Their scope is focused on the safety of the trial, using sets of data obtained in pre-specified intervals. They should have a pre-planned statistical analysis capable of detecting both benefits and harms occurring in one arm of the study. The US FDA has issued rules for the operation of DSMBs25. Reasons to stop a trial are futility (e.g., a meager chance that adding more patients may prove the hypothesis or could change the conclusion of the study), low accrual rates, high non-compliance, or one of the groups experiencing benefit or excessive risk. DSMBs are authorized to propose changes in the protocol to decrease the risk of adverse events (e.g., by changing selection criteria or the dosage of the study product, or by including an additional test).
LACK OF TRANSPARENCY IN THE PLANNING AND EXECUTION OF THE STUDIES
The complex nature of the Pharma-sponsored studies limits the participation of the primary investigators (even for those in the Steering Committee). Sponsors are the only ones that have full access to the study information. Although papers are presented and authored by independent investigators, statistical analyses are performed by Pharma personnel and manuscripts are prepared by medical writers26.
Tools to overcome the problem
RECs should request that all investigators disclose their conflict of interest related to the sponsor or the study. Sponsors should disclose the functions of each member of the steering committee and the individuals responsible for each one of the critical steps of the study.
CONCLUSIONS
Pharma-sponsored clinical studies are among the most complex scenarios that RECs may face. Institutional, economic, or personal motivations to participate in these initiatives add pressure to the already complex work of the RECs. Changes in the selection criteria or the study design requested by a local REC may exclude the research center from the initiative. However, RECs should keep in mind that they are the first line of protection against low-quality or high-risk studies driven by economic interests. In this manuscript, we have reviewed the most common problems found in Pharma-sponsored studies. Potential solutions are suggested to prevent or clear up some controversial issues (Fig. 2). Even so, different challenges will be faced soon, when new therapies (i.e., new forms of genome editing or genetic engineering) reach their late stage of development27. On the other hand, Pharma-sponsored research is a crucial driver to develop and implement innovative approaches to improve the informed consent process or the execution of studies. All parties involved in research should work together to guarantee the protection of participants, the paramount principle on which clinical investigation is based28.


