











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
In the medical literature of the 20th century, there are isolated cases of patients reported with clinical and electrocardiographic (ECG) characteristics of what is now known as catecholaminergic polymorphic ventricular tachycardia (CPVT);1,2 nevertheless, it was not until 1975 that Reid et al3. reported a typical case of the disease. They presented a young girl (6-year-old) without cardiac structural abnormality, with bidirectional ventricular tachycardia precipitated by effort and emotional stress. However, Coumel et al. were the first to gather a case series and introduce the term catecholamine-induced4. With this cumulative series (4 cases of their own and 10 more from the literature) they provided the basis to consider CPVT as an independent entity. Almost 20 years later, in 1995, the same French group5 enlarged their series to 21 patients and could characterize the arrhythmia as consisting of isolated polymorphic ventricular extrasystoles followed by salvoes of bidirectional and polymorphic tachycardia susceptible to degeneration into ventricular fibrillation (VF). Again, its reproducibility by any form of increasing adrenergic stimulation, as previously known, was a common denominator. As there was a familial history of syncope or sudden death in 30% of the patients, some groups named the disease as familial polymorphic ventricular tachycardia. β-blocker therapy usually resulted in control of symptoms and tachyarrhythmias; further developments of syncopal events or unexpected deaths were attributed to treatment interruption. It was not until 2001 that Priori et al6. and Laitinen et al7. provide us with a better understanding of the molecular pathogenic mechanism through the study of the isoform two of the intracellular Ca2+ channel/ryanodine receptor (RYR2), which is the major cellular mediator of a calcium-induced calcium release (CICR) mechanism in animal cells. Although mutations in five different genes (RYR2, CASQ2, CALM1, TRDN, and TECRL) are associated with CPVT phenotype, RYR2 missense mutations are implicated in up to 60 % of all CPVT cases8,9.
In Mexico, a recent estimate reports a prevalence of 9.5/1000 patients/year for sudden death in children10; unfortunately, there is no specific data in the country for the prevalence of CPVT in children. On the other hand, CPVT is estimated in 1/10,00011 with sudden death as the first presentation in <30% of the cases12. This review highlights insights from clinical, molecular, and genetic features of CPVT.
CLINICAL DIAGNOSIS
Diagnosis of CPVT must be suspected in individuals, especially children, with exercise- or emotion-induced syncope13. Affected subjects may have a family history of sudden juvenile death or stress-induced syncope5, although this is not a rule14. Patients may present life-threatening VT or VF during emotional or physical stress, with exercise-related syncope; even sudden cardiac death could be the first manifestation of the disease13,14. The diagnosis could be so difficult that in the series of 21 patients described by Leenhardt et al., near half of the children were initially considered with epilepsy and treated with anticonvulsants for several months and in some cases, years5.
In the most-recent registry of 27 pediatric centers, including 226 patients evaluated as part of the family screening, 78% presented with symptoms, and 19% were asymptomatic. Syncope was the most frequent presentation occurring in 64%, followed by nonfatal cardiac arrest in 33%, palpitations in 7%, and chest pain in 4%. The median age of onset of symptoms was 10.8 years (6.8-13.2). As previously mentioned, in 38% of patients there was a delay in the diagnosis for more than 1 year, and 56% of children initially received a misdiagnosis15.
Two main ECG types of polymorphic VT have been described16, the polymorphic VT and the bidirectional tachycardia (with electrical alternans of QRS). Typically, the arrhythmia begins above a heart rate threshold of 120-130 bpm, initially with isolated monomorphic and then polymorphic ventricular extrasystoles; if the activity is stopped, the arrhythmia disappears without clinical consequences. If the effort continues, PVC is followed by salvoes of bidirectional and then polymorphic VT (350-400 bpm) susceptible to degeneration into VF5.
To exemplify the clinical variability of CPVT, the following examples are relevant. In a case series where 14 of the 30 probands had RYR2 mutations, the affected patients had events at a younger age (first syncope at 8 vs. 20 years), and male sex was a significant risk factor for syncope compared to those without such mutations. In contrast, 90% of the subjects without an RYR2 mutation were female. The arrhythmias documented in those 14 patients were bidirectional VT in 36%, polymorphic VT in 58%, and catecholaminergic idiopathic VF in 50%13. In the case of genomic deletion in RYR2 and polymorphic VT, other clinical manifestations have been observed including sinoatrial node and atrioventricular node dysfunction, atrial fibrillation, and dilated cardiomyopathy17. In a second genetic form of CPVT, due to mutations in CASQ2, the first seven affected families included children who died suddenly at an average age of 7 years and 12 of 41 family members with a history of recurrent syncope or seizures beginning at 6 years of age. All the 12 symptomatic patients had relative resting bradycardia and presented polymorphic VT induced by exercise18.
Involvement of RYR2 Missense Mutations in the Arrhythmogenic CPVT Disorder
Calcium in ionic form (Ca2+) is an essential and intracellular messenger of cardiac contractile cells (cardiomyocytes) due to its significant participation in several cellular processes, including gene expression, cell differentiation, apoptosis, and cell contraction; the latter was stated for the first time by Sidney Ringer19. Periodic fluctuations in the intracellular Ca2+ concentration ([Ca2+]i) of cardiomyocytes control the magnitude and duration of contractile force developed by the cardiac cells20. Under physiological conditions, cardiomyocytes translate action potentials into mechanic force through the excitation-contraction coupling (ECC) mechanism. In the ECC, the sarcolemma depolarization activates voltage-dependent L-type Ca2+ channels. Located at the T-tubules, they induce an inward Ca2+ current (ICa) that, in turn, elicits the release of more Ca2+ through the aperture of clusters of RyRs, through the CICR mechanism20 (Fig. 1). Hence, the RyR2 has a key role in the ECC as the cation channel responsible for the massive release of Ca2+ from the sarcoplasmic reticulum (SR) in the cardiac systole.
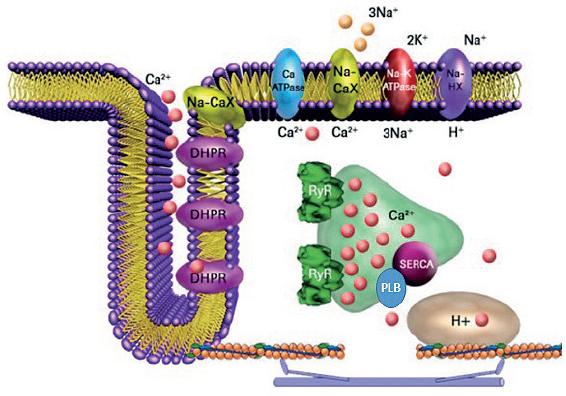
Figure 1 Cardiac excitation-contraction coupling. Schematic representation of a T-tubule in a ventricular cardiomyocyte. Ca2+, calcium in its ionic form; DHRP, dihydropyridine receptor or L-type Ca2+ channel; Na-CaX, sodium/calcium exchanger; PLB, phospholamban; SERCA, sarco/endoplasmic reticulum Ca2+ pump; RyR, ryanodine receptor; H+, proton gradient in mitochondria.
However, it is recognized that RyRs are not entirely closed during the relaxation phase of the cardiac cycle. In diastole, the RyRs release Ca2+ in several forms mainly as Ca2+ sparks, Ca2+ wavelet, and Ca2+ waves giving basis to a diastolic Ca2+ leak. In physiological conditions, RyRs participate in the regulation of SR Ca2+ load21-24. However, under pathological conditions, abnormal diastolic Ca2+ leak through RyR2 participates in the generation of both acquired and hereditary arrhythmias, such as CPVT25.
Although the gene that codifies for RyR2 has only one copy in the human genome located in chromosome 1, locus 1q4326 it contains 105 exons and is considered one of the largest genes found in our genome27. With 4967 residues per monomer, the functional hRYR2 (homotetramer) is the largest ion channel currently known. Since 2001, more than 150 RyR missense mutations have been associated with the CPVT phenotype (human gene mutation database; http://www.hgmd.cf.ac.uk/ac/index.php). The RyR2 isoforms are originated by missense mutations occurring mainly at four hotspots: N-terminal (CPVT1-I, RyR2 residues: 62-466); central (CPVT1-II, RyR2 residues: 2246-2534), C-terminal (now CPVT1-IV, RyR2 residues: 4497-4959), and the recently defined hotspot-3 (CPVT1-III, RyR2 residues: 3778-4201)28. Due to the lately described near-atomic resolutions of the RyR2 from the porcine heart in its open and closed conformations29, it is possible to locate these CPVT hotspots into distinct structural domains (Fig. 2). For instance, the CPVT1-I is in the N-terminal domain of the protein. The CPVT1-II is delimited inside the HD1 domain. The CPVT1-III, even though it is absent in the RyR1 isoform, encompasses the central domain and the U motif of RyR2. Finally, the CPVT1-IV hotspot is in the channel domain. Mutations occurring in CPVT1-III hotspot are currently of relevant interest due to their localization at the central domain of the RyR2 (Fig. 3). The central domain serves as the primary transducer that integrates and translates the conformational changes of the cytoplasmic domains to channel gating28, evidencing that alterations in intracellular Ca2+ handling are involved in the generation of arrhythmias8,30. Furthermore, mutations at CPTV1-III and -IV may also have a profound impact on the cytosolic Ca2+ activation of RyR2, and on the actions of modulators such as caffeine and ATP, since they could be in the boundaries of binding sites for these ligands (Fig. 3).
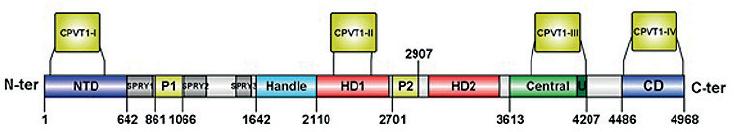
Figure 2 Localization of catecholaminergic polymorphic ventricular tachycardia (CPVT) hotspots in RYR2 structural domains. Linear representation of structural domains identified in the RYR2 monomer. The domain boundaries are assigned according to Peng et al29. The four CPVT1 hotspots (CPVT1-I to IV) are depicted above the domains. NTD, N-terminal domain; SPRY1-3, P1, and P2 domains; Handle, Handle domain; HD1-2, helical domains 1 and 2; U, U motif; CD, channel domain. Scheme prepared with IBS illustrator84.
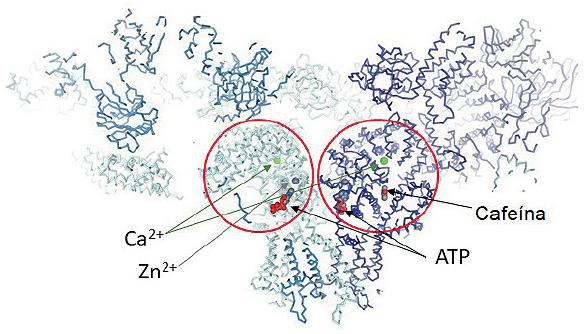
Figure 3 Representation of two RyR monomers bound with four different ligands: Ca2+ (green balls), Zn2+ (gray balls), ATP and caffeine, (PDB accession code 5TAL). The red circles highlight the Central and Channel Domain of the RyR where the ligands and most of the catecholaminergic polymorphic ventricular tachycardia 1-III and IV mutations are found.
Only a few numbers of RyR2 variants have been characterized at the molecular level; most of them generate ion channels with a gain-of-function that lead to out of control diastolic Ca2+ leak27,31. The pathological consequence of an augmented diastolic Ca2+ leak is that it would activate the Na+/Ca2+ exchanger generating a net inward current that is causative of delayed afterdepolarizations (DADs), triggering additional RyR2 activation, and aftercontractions32.
One of the first RyR2 point mutations related to a dominant form of CPVT was R4496C6,33, and a knock-in transgenic mouse harboring this mutation was generated34. The mice presented an arrhythmogenic phenotype very similar to the clinical manifestations of patients carrying the RyRR4497C including the presence of bidirectional VT. Furthermore, after challenging the RyRR4497C cardiomyocytes with both adrenaline and caffeine, the cells show DADs, suggesting that Ca2+-dependent arrhythmias are elicited by β-adrenergic activation. Intact quiescent RyRR4497C cardiomyocytes exhibit increased occurrence of Ca2+ sparks and Ca2+ waves indicative of an abnormal diastolic Ca2+ leak. This augmented Ca2+ release is further enhanced by β-adrenergic stimulation and increasing frequency, similarly to what happens in humans with CPVT35.
This scheme is adequate for most RyR2-linked CPVT mutations. However, in 2002, Priori et al. also reported a special RyR2-A4860G mutation12. When the recombinant RyR2-A4860G channels were expressed in HEK293cells, one striking feature of the RyR2-A4860G channels was the loss of luminal Ca2+ sensitivity that generates loss-of-function channels36; then the question was, how do hypoactive channels participate in cardiac arrhythmias? Thus, the group of Valdivia et al. generated a transgenic mouse harboring the RyR2-A4860G channel with loss-of-function that exhibited basal bradycardia, but after sympathetic stimulation, the mice developed malignant arrhythmias37. At a single-cell level, the loss-of-function mutations are related to diminished Ca2+ activation and progressive SR Ca2+ overload9,38 that may lead to abnormal SR Ca2+ release and subsequent activation of NCX. Other examples of these mutations are I4855M, S4938F, and E4872Q38,39. Interestingly, all of these mutations are located mainly on the channel domain where the Ca2+ binding site is situated40 (Fig. 3).
These novel findings have profound therapeutic implications since CPVT treatment should not assume that CPVT-linked RyR2 mutations produce gain-of-function channels only; thus, future work should also focus on developing more suitable therapeutic interventions for patients with arrhythmias associated to loss-of-function channels.
Mechanisms of Arrhythmogenesis in Other Affected Genes: calsequestrin, Calmodulin, and Triadin
ECC mechanism relies on the right arrangement of the dyads and the synchronous activity of the Ca2+ release unit (CRU) proteins; during this process, the opening of RyR2 will allow the massive Ca2+ release from the SR, the increase in the free cytoplasmic Ca2+ and the cellular contraction. At the luminal side, the RyR2 is tightly regulated by some proteins that directly interact with it and modify its Ca2+ sensitivity and its kinetics. These proteins, named calsequestrin (CASQ), triadin (TRD), junctin, and HRC, are critical regulators of RyR2 and endow the channel with an excellent fine tuning41,42. At the cytosolic side, at least seven different accessory proteins bind the receptor, but we will mention only calmodulin (CALM). Any alterations to these proteins will be translated into functional changes in the Ca2+ regulatory machinery and finally in an alteration in the intracellular Ca2+ homeostasis.
About these proteins related to CPVT, there are at least 14 mutations in the gene encoding for cardiac calsequestrin (http://triad.fsm.it/cardmoc/), 3 in the gene encoding for triadin43, and 3 more for calmodulin44,45. These mutations will result in an altered structure of the protein by a) modifying its Ca2+ binding properties, as occurs with CALM1 A53I and A97S or CALM3 A103V mutants42 or with the CASQ2 R33Q46; b) making the protein readily degradable as CASQ2 R33Q or TRD T59R43,45,47; or c) decreasing its expression by producing a non-functional protein due to the presence of premature stop codons47,48. Independent of the mechanism, the Ca2+ release from SR is altered thus inducing the CPVT phenotype.
CPVT has been considered a cardiomyopathy with no structural heart alterations; however, in 2006, Knollman et al. engineered a CPVT2 animal model, the CASQ2-null mouse, and demonstrated for the 1st time that anatomical heart abnormalities in CPVT are not evident, since structural changes are inside cardiomyocytes, in the structure of the dyads, specifically in the junctional SR49. In experiments performed in CPVT2 animal models, the absence or decrease in expression of calsequestrin induces ultrastructure alterations of the heart, namely disarray on the coupon due to an increase in the widening of the JSR, a decrease in the number of coupons, and increase in the JSR volume46,49,50. The ultrastructural abnormalities can be long-lasting and reversed if exogenous Casq2 is expressed in the cardiac cells50,51. Similar results were obtained in the Triadin null mouse, where it was also demonstrated that the absence of triadin decreases the extent of JSR by reducing the length and number of contacts between JSR and T tubules; furthermore, the Cav1.2 channels juxtaposed to RyR2 and the colocalization between CASQ2 and RyR2 were decreased without changes in the expression of Cav1.252,53.
Interestingly, the absence of TRD or CASQ2 is accompanied by a decrease in expression of CASQ2 or TRD, respectively46,49,50,52. Furthermore, other CRU proteins such as RYR2, junctin or junctophilin 2 are also downregulated or upregulated in some animal models54. These experiments highlight, first, the crucial role of triadin and calsequestrin in maintaining the architecture of the dyad and the effectiveness of the ECC and second, the fact that both proteins are vital for the heart function.
Regarding calmodulin, in 2012 Nyegaard et al., reported two missense mutations in CALM1. The N53I was found in a Swedish family with the dominant form of CPVT-like arrhythmias, while the other mutant, N97S, was identified in an Iraqi-origin woman diagnosed with CPVT44. A third mutation, A103V, this time in the isoform 3 of CALM, was found in a woman clinically diagnosed with CPVT45. The three mutations lead to altered calcium binding properties of calmodulin, and only the CALM1 A97S also showed a deficient interaction with the RYR2 calmodulin binding domain at low calcium concentration (< 100 nM)44. In vitro, functional evaluation revealed that all the mutants promoted calcium sparks and a decrease in the SR calcium content. CALM1 N97S and CALM3 A103V also decreased the calcium-dependent inactivation of Cav1.2 with no significant effects on the action potential morphology45.
Finally, the animal models of CPVT in which the effect of several mutant proteins has been characterized, have advanced the knowledge on the pathophysiology of CPVT, i.e., (1) the widening of JSR is a compensatory response to the decreased capacity of CASQ2 in the SR to store calcium. (2) A disarray in JSR around T tubules directly affects the coupons and the effectiveness in communication between voltage-gated calcium channels in the plasma membrane and RYR2 in the SR membrane, therefore, affecting downstream the entire ECC. (3) Since triadin and junctin work in binding CASQ2 to ryanodine receptor, the altered interaction between these proteins will decrease the RyR2-calcium modulation, inducing calcium leakage from JSR. (4) CALM binding to RyR2 decreases the open probability of the channel, and a decrease in the calmodulin sensitivity to calcium or a defective interaction with the RyR2 could induce a leaky ryanodine receptor by increasing opening conformation of the channel. (5) Continuous SR calcium leakage increases the risk of spontaneous calcium release and CPVT.
Involvement of TECRL Protein in CPVT
The trans-2,3-enoyl-CoA reductase-like (TECRL) protein does not have a direct effect on the cardiac phenotype, although pluripotent stem cell-derived cardiomyocytes have been shown to have a participation in the action potential duration. In addition, it has been observed that knocking of TECRL induces a decrease of RyR2 and CASQ255. This gene has recently been located in 4q13.1 locus, has 12 exons, and spans 134 kb (online mendelian inheritance in man [OMIM], http://omim.org/entry/617242). Two studies have reported a mutation in one of six affected children carrying a homozygous splice site mutation c.331+1G > A56 and, in two French-Canadian women, a homozygous mutation R196Q55.
Genetic Testing
Genetic testing has an essential role in diagnosis, management, pre-symptomatic diagnosis, counseling and treatment of the proband, and in some cases the information obtained can be useful for offspring and relatives57,58. In CPVT, there are five genes identified with point mutations or deletions associated to CPVT phenotype7,33,43,44,47,48,59,60, being RYR233,59, CALM144, TRDN47, and CASQ60 the affected genes. In this group, the gene with most mutations related to CPVT is RYR2, and together the five genes can explain 60% of cases of CPVT in patients with recessive or dominant inheritance, failing to account for 40% of cases with CPVT61. The increase in association between genes affected in CPVT has made genetic screening part of the tools for risk stratification in cardiogenetic diseases. At present, there are five molecular types described in OMIM (OMIM, http://www.omim.org) for CPVT62 (Table 1). Type 3 has been delimited recently by linkage of the marker D7S493, associated with the disease in one endogamic family56, and after functional studies, the affected gene was found to be TECRL; a knockout in vitro model for this gene showed a reduction of the proteins RYR2 and CASQ255,63. All of these five genes have diverse characteristics, for example, different sequence size, number of coding and no coding sequences, promoters, repressors, enhancers, splicing, and protein isoforms64, which could complicate the molecular study and interpretation.
Table 1 Molecular classification of CPVT by OMIM*.
| Type | Mendelian pattern | Gene | Locus | Most common clinical data | Miscellaneous |
|---|---|---|---|---|---|
| CPVT Type 1 OMIM (#604772) | Autosomal Dominant | RYR2 | 1q43 | Polymorphic ventricular tachycardia induced by physical activity, stress, or catecholamine infusion, syncope, sudden death | Sinoatrial node dysfunction, atrioventricular node dysfunction, atrial fibrillation, atrial standstill left ventricular dysfunction left ventricular dilation, seizures |
| CPVT Type 2 OMIM (#611938) | Autosomal Recessive | CASQ2 | 1P13.1 | Polymorphic ventricular tachycardia induced by physical activity, stress, or catecholamine infusion, bradycardia, relative resting, syncope, sudden death | Seizures |
| CPVT Type 3 OMIM (#614021) | Autosomal Recessive (consanguineous family) | Marker D7S493 (TECRL) | 7p22-p14 | Polymorphic ventricular tachycardia induced by physical activity, borderline prolonged QT interval, syncope, sudden death | Onset at an early age, associated with sudden death in childhood |
| CPVT Type 4 OMIM (#614916) | Autosomal dominant | CALM1 | 14q32.11 | Polymorphic ventricular tachycardia induced by physical activity or stress, dizziness, syncope, cardiac arrest, sudden death, prominent U-waves in anterior leads on electrocardiogram, premature ventricular contractions, including couplets and triplets of variable morphology | Onset within the first decade of life |
| CPVT Type 5 OMIM (#61544) | Autosomal recessive | TRDN | 6q22.31 | Polymorphic or bidirectional ventricular extra systoles polymorphic ventricular tachycardia exercise-induced syncope | Proximal muscle weakness, cardiac arrest, and sudden death may occur, even in early childhood |
*Modified from OMIM: Online Mendelian Inheritance in Man (Marla J. F. ONeill: December 28, 2011), CPVT: catecholaminergic polymorphic ventricular tachycardia
Different types of techniques for DNA testing: A genetic testing, in essence, is accurate, with low cost, the results may be quickly obtained, and the technique used has a wide coverage for the affected genes. As the first option, the classical approach has been the study of selected exons, deletion/duplication analysis or direct sequence analysis based on the polymerase chain reaction65 and subsequent sanger sequencing66 (first-generation sequencing). Limiting factors of these conventional molecular techniques include low coverage, lack of amplification of large fragments or whole genes in a single assay, and also are time consuming; due to the genetic heterogeneity (five genes are identified). A recent solution is the use of massive parallel sequencing (Second-generation sequencing), where millions of small copies of DNA fragments are simultaneously amplified and sequenced. There are different variants of massive parallel sequencing, but together they are also known as next-generation sequencing (NGS). Due to the high coverage of NGS and the genetic heterogeneity in CPVT, different companies have developed sequencing panels including part of or all the five genes described for CPVT. An alternative to gene panels, in negative cases there is the possibility to study the suspected case through whole genome sequencing or whole exome sequencing (WES)61. In WES, the sequenced bases cover 1% of the genome, and this percentage belongs to all the coding sequences in an individual; with both techniques, new mutations related to CPVT have been found. However, there could be an associated overrepresentation of a mutation in the large whole genome and exome databases; due to this, the clinical and molecular biologist must be cautious with the results, being necessary to validate in vitro, in vivo67 and supported by at least four bioinformatic analysis.
A drawback of these new techniques is the significant amount of data and the need for more sophisticated tools and mathematical modeling, as well as the obtention of unsolicited findings related to diseases other than CPVT, which implies ethical issues for patients, offspring, relatives, and the medical team, including the cardiologist and clinical geneticist67,68.
Indications for genetic testing: the Heart Rhythm Society (HRS) in collaboration with the European Heart Rhythm Association (EHRA), elaborated a consensus statement for genetic testing in channelopathies, recommending the genetic study as Class I for RYR2 and CASQ2 for patients with a high suspicion of CPVT based on clinical, electrophysiological, and functional studies. If a mutation in an index case is found, cascade screening is recommended69,70. However, if the primary RYR2 genetic screening is negative, a phenocopy should be considered. If ectopy is present and abundant on 24-h ambulatory monitoring and prominent U wave, it is mandatory to consider KCNJ2 testing for Andersen-Tawil Syndrome69. Due to the recent discovery of TRDN47, CALM144, and TECRL55 genes as causes of CPVT, their inclusion in previous guidelines had not been contemplated, but we recommend screening after exclusion of RYR2 and CASQ as causes of this disease. Other genes to consider in a genetic test are KCNJ271-74 and ANKB75.
Treatment
The participation in competitive sports and strenuous exercise increases the risk of ventricular arrhythmias in patients with CPVT due to the rise in catecholamines associated with exertion. As such, it is recommended that patients with CPVT avoid competitive sports13. Patients who have sustained ventricular arrhythmias are treated with an implantable cardioverter defibrillator (ICD) to terminate arrhythmias and reduce the risk of syncope and/or sudden cardiac death. However, patients should also receive medical therapy in addition to the ICD in an effort to prevent arrhythmias and minimize ICD shocks76.
β-blockers are the cornerstone of treatment due to the catecholaminergic nature of the arrhythmias. Long-acting β-blockers without intrinsic sympathomimetic activity, such as nadolol, are preferred for all patients with spontaneous or documented stress-induced ventricular arrhythmias77. Unfortunately, nadolol is not available in all countries (e.g., Mexico). In the most extensive reported series, 101 patients with CPVT followed for a mean of 7.9 years, β-blockers were administered to 81 patients (80%) immediately after diagnosis. Of those taking a β-blocker, 62 (77 %) experienced no events during follow-up, and 9 (11%) experienced fatal or near-fatal events. At least 4 of these 9 patients did not take their β-blocker on the day of the event14. In a systematic analysis of 11 observational studies including 403 patients with CPVT, 18.6% and 37.2 % of the arrhythmic events (syncope, aborted cardiac arrest, and sudden cardiac death) occurred at 4 and 8 years, respectively77.
In patients with ongoing arrhythmias despite therapy with a β-blocker, the addition of flecainide may be useful for suppressing ventricular arrhythmias55,78. Flecainide is best known as a blocker of cardiac sodium channels. However, it also inhibits the cardiac ryanodine receptor (RYR2). In a report of two patients, the addition of flecainide prevented further episodes of CPVT; interestingly, this effect persisted in one patient after the withdrawal of the β-blocker and calcium channel blocker79. A subsequent series of 10 patients with CPVT type 2 who had recurrent VT despite therapy with a β-blocker (all patients) and calcium channel blockers (in 6 of 10 patients), found flecainide to be effective in suppressing exercise-induced ventricular tachyarrhythmia when given in addition to β-blockers80.
In a study of 24 patients with CPVT who underwent ICD implantation and were followed for a median of 3.3 years, 14 patients (58 %) received a total of 140 shocks, of which approximately 54 % were felt to be adequate shocks81. Some groups propose they left cardiac sympathetic denervation (LCSD) for patients with CPVT who remain symptomatic after maximal medical therapy. Data from small case series suggest a role for LCSD in patients with CPVT82. Among the most extensive reported series, there is one of 63 CPVT patients treated with LCSD who were followed for 37 months; of 38 patients with arrhythmias while on optimal medical therapy, only 12 patients (32%) had a recurrent arrhythmic event following LCSD83.
Recommendations from the 2013 HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes for the management of CPVT are summarized in Table 2.
Table 2 Expert consensus recommendations on CPVT therapeutic interventions
| Class I | 1. Recommended lifestyle changes a) Limit/avoid competitive sports b) Limit/avoid strenuous exercise c) Limit exposure to stressful environments 2. β-blockers are supported in all symptomatic patients 3. ICD implantation is recommended in patients who experience cardiac arrest, recurrent syncope, or polymorphic/bidirectional VT despite optimal medical management, and/or LCSD |
| Class IIa | 4. Flecainide can be a useful addition to
β-blockers in patients who experience recurrent syncope or
polymorphic/bidirectional VT while on β-blockers 5. β-blockers can be helpful in carriers of a pathogenic CPVT mutation without clinical manifestations of CPVT |
| Class IIb | 6. LCSD may be considered in patients who experience recurrent syncope or polymorphic/bidirectional VT/several appropriate ICD shocks while on β-blockers and in patients who are intolerant or with contraindication to β-blockers |
| Class III | 7. ICD as a standalone therapy is not indicated in
an asymptomatic patient with a diagnosis of CPVT 8. Programmed electrical stimulation is not indicated in CPVT patients |
Modified from Priori et al13. CPVT: catecholaminergic polymorphic ventricular tachycardia, ICD: implantable cardioverter defibrillator, LCSD: left cardiac sympathetic denervation
CONCLUSIONS
CPVT is a disease that conveys a risk of sudden death in young people; therefore, its correct identification and diagnosis at an early stage can prevent fatal consequences. Although CPVT was identified since 1975 and its molecular characterization dates back to 2001, it is only in recent years, that an increased number of cases have been genetically confirmed thanks to NGS.

