










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de investigación clínica
On-line version ISSN 2564-8896Print version ISSN 0034-8376
Rev. invest. clín. vol.58 n.2 Ciudad de México Mar./Apr. 2006
Artículo de revisión
Fibrosis quística: la frontera del conocimiento molecular y sus aplicaciones clínicas
Cystic fibrosis: molecular update and clinical implications
Lorena Orozco,* Margarita Chávez,** Yolanda Saldaña,* Rafael Velázquez,* Alessandra Carnevale,*** Ariadna González–del Ángel,**Silvia Jiménez*
* Laboratorio de Genómica de Enfermedades Multifactoriales, Instituto Nacional de Medicina Genómica, S.S.
** Laboratorio de Biología Molecular, Departamento de Investigación en Genética Humana, Instituto Nacional de Pediatría, S. S.
*** Subdirección General Médica del ISSSTE.
Reimpresos:
Dra. Lorena Orozco
Laboratorio de Genómica de Enfermedades Multifactoriales Instituto Nacional de Medicina Genómica
Periférico Sur No. 4124, Torre Zafiro 2, Piso 5
Del. Alvaro Obregón
01900, México, D.F.
Tel: + 55 53–50–19–57 Fax: + 55 53–50–19–50
Correo electrónico: lorozco@inmegen.gob.mx
Recibido el 8 de marzo de 2005.
Aceptado el 4 de octubre de 2005.
ABSTRACT
Cystic fibrosis (CF) is an autosomal recessive disorder characterized by chronic pneumopathy, pancreatic insufficiency, elevated sweat chloride levels and male infertility. It is caused by defects in the CF trans membrane conductance regulator (CFTR) gene, which encodes a protein that functions as a chloride channel. The identification of the CF–causing gene was a landmark in molecular medicine. Currently, over 1,300 disease–causing mutations have been reported to the Cystic fibrosis genetic analysis consortium. ÁF508 mutation is the most common CF alíele, however a high heterogeneity of the CFTR mutations spectrum has been observed in populations, particularly in southern Europe and Latin America. Depending on the effect at the protein level, CFTR mutations can be divided in at least 5 classes. These mutations could cause totally (classes I–III) or partially (classes IV and V) loss of the protein function. The molecular defects resulting from different mutations in CFTR partially explain the clinical heterogeneity of the disease, suggesting the existence of modifier genes that are involved in modulating the phenotype and severity of the CF. In this review, we discuss the fundamental aspects and the recent progress that could give to the lector, the knowledge to understand the CFTR gene structure, the function of the CFTR protein, how CF mutations disrupt it, its phenotype consequences and finally, the strategies to design new therapies for the disease.
Key words. Cystic fibrosis. Cystic fibrosis transmembrane conductance regulator. Mutation classes. Genotype. Phenotype.
RESUMEN
La fibrosis quística (FQ) es un padecimiento autosómico recesivo que se caracteriza por neumopatía crónica, insuficiencia pancreática, elevación de cloruros en sudor e infertilidad masculina. Esta patología es causada por la presencia de mutaciones en el gen CFTR que codifica para un canal de cloro denominado proteína reguladora de la conductancia transmembranal (CFTR). Hasta la fecha se han reportado alrededor de 1,300 mutaciones diferentes, cuya frecuencia varía entre los diversos grupos étnicos. Estas mutaciones condicionan la pérdida total (clases I, II y III) o parcial (clases IV y V) de la función de la proteína y causan un defecto en el transporte de electrólitos en la membrana apical de las células epiteliales. Con excepción de la función pancreática, las manifestaciones clínicas de la FQ son variables aun en pacientes con el mismo genotipo, por lo que la presencia de las diferentes mutaciones en el CFTR explica sólo parcialmente la heterogeneidad clínica de la FQ. Recientemente se ha propuesto que otros genes denominados genes modificadores participan en la gravedad del cuadro clínico. Así, la FQ es una enfermedad genética que resulta en un amplio espectro de manifestaciones clínicas que pueden ir desde muy leves hasta conducir a la muerte durante los primeros meses de vida, por lo que en algunos casos el diagnóstico es sumamente complejo. En los últimos años, el gran alud de conocimientos ha permitido entender el defecto básico de la enfermedad y los mecanismos que la condicionan, por lo que en esta revisión se discuten los fundamentos para el entendimiento de la fisiopatología de la FQ, desde los aspectos clínicos hasta los avances moleculares más recientes.
Palabras clave. Fibrosis quística. Proteína reguladora de la conductancia transmembranal de la fibrosis quística. Clasificación de las mutaciones. Genotipo. Fenotipo.
INTRODUCCIÓN
La fibrosis quística o mucovisidosis (FQ; OMIM #219700) es el padecimiento autosómico recesivo más frecuente en la población caucásica, donde se presenta con una incidencia de uno en cada 2,000–3,500 recién nacidos vivos y se estima que uno de cada 25 individuos es portador sano de la mutación.1 En los países desarrollados los pacientes con fibrosis quística tienen una expectativa de vida de aproximadamente 35 años, mientras que en el nuestro es de ocho años.2
Este padecimiento es ocasionado por mutaciones en el gen CFTR, el cual codifica para una proteína transmembranal cuya función más importante es la de canal de cloro (Cl-). La pérdida de la función de esta proteína causa un defecto en el transporte de electrólitos en la membrana apical de las células epiteliales alterando la función secretoria en diferentes órganos y tejidos. Este defecto conduce a la producción de secreciones exocrinas anormalmente viscosas y conlleva a una enfermedad multisistémica y progresiva con expresividad muy variable, la FQ.3 En la década de los 90s, la clonación del gen responsable permitió un enorme progreso en el conocimiento de la función de la CFTR y en la caracterización de las mutaciones responsables de la enfermedad, cuya repercusión en el manejo integral del paciente con FQ no tiene precedente. Sin duda, estos avances han conducido a mejorar las estrategias preventivas, de diagnóstico y de tratamiento, por lo que en esta revisión se proporcionan los fundamentos para el entendimiento de la fisiopatología de la enfermedad, desde la clínica hasta los aspectos moleculares más recientes.
ALTERACIONES PATOLÓGICAS Y MANIFESTACIONES CLÍNICAS
El signo más consistente de la FQ es la concentración elevada de Cl-, Na+ y K+ en la secreción de las glándulas sudoríparas. La concentración de Cl- puede ser medida por el iontoforómetro después de la estimulación con pilocarpina, donde valores por arriba de 60 mEq/L son sugestivos de FQ.4 La pérdida excesiva de agua y electrólitos puede ocasionar alcalosis hipoclorémica e hiponatrémica, sobre todo en presencia de vómito, diarrea o exposición prolongada al sol.
La afección del tracto respiratorio es la manifestación clínica más grave en esta entidad. Inicialmente se manifiesta con tos intermitente que deriva en inflamación crónica provocada por secreciones mucosas espesas e infecciones recurrentes por microorganismos oportunistas, como Pseudomona aeruginosa y Staphylococcus aureus. Este proceso genera bronquiectasias que conducen a un cuadro obstructivo–restrictivo de las vías respiratorias con hipertensión arterial pulmonar y posteriormente corpulmonale.5 Colateralmente, las lesiones inflamatorias crónicas de la mucosa generan pedunculaciones y formación de pólipos nasales. La afección respiratoria y las infecciones persistentes son las principales causas de muerte entre la primera y la cuarta décadas de la vida.6
Otras manifestaciones relevantes en FQ son las alteraciones que comprometen al tracto gastrointestinal y glándulas anexas. El 85% de los pacientes sufre de insuficiencia pancreática (IP) y el resto muestra una función pancreática suficiente (SP) para permitir una digestión normal. La clasificación de los pacientes se basa en la presencia (IP) o ausencia (SP) de esteatorrea secundaria a la mala digestión y mala absorción de las grasas. La IP puede conducir a desnutrición y cuadros carenciales de vitaminas A, D, E, K, cinc, etc.7,8 En contraste con la afección pulmonar, el páncreas muestra una respuesta inflamatoria muy pobre. Los hallazgos patológicos pueden ser evidentes desde el último trimestre de la gestación y conforme la dilatación de los acinos progresa, la fibrosis del páncreas se hace más aparente y en estados más avanzados el tejido pancreático es reemplazado por tejido adiposo y fibroso. De hecho, la apariencia patológica distintiva de este órgano condujo a Dorothea Anderson a denominar esta enfermedad "fibrosis quística del páncreas".9 En la segunda década de la vida la función endocrina del páncreas se encuentra afectada en 27–42% de los pacientes, aunque sólo 8% de los casos desarrolla diabetes insulino–dependiente. Otra alteración del aparato digestivo es la afectación hepática que se ha encontrado en 25% en las autopsias de los pacientes con FQ, pero sólo de 2 a 15% desarrolla cirrosis biliar.10 Aproximadamente 20% de los recién nacidos con FQ presenta íleo meconial causado por el acumulo de meconio en la región distal del intestino, probablemente por insuficiencia enzimática temprana.1,11
Por otra parte, aproximadamente 99% de los varones con FQ presenta infertilidad por azoospermia, con alteración de las estructuras wolfianas y la consecuente obstrucción, atrofia o ausencia de los conductos deferentes, epidídimo y vesículas seminales. Estos cambios anatómicos explican la ausencia de espermatozoides durante la eyaculación, a pesar de que en estos pacientes existe una espermatogénesis normal.12 Por otra parte, la esterilidad por azoospermia obstructiva aislada puede ser la expresión mínima de una FQ atípica, por lo que esta entidad debe considerarse en el diagnóstico diferencial de cualquier individuo con esterilidad por azoospermia obstructiva. En las mujeres con FQ la esterilidad es poco frecuente (3–5%) y puede deberse a alteraciones en el moco cervical, a amenorrea o a ciclos de anovulación debidos a la gravedad del cuadro clínico o al estado nutricional.12 En las pacientes fértiles la mayoría de los embarazos ha tenido un curso normal.
HERENCIA
La FQ tiene un modo de herencia autosómico recesivo, afecta a ambos sexos por igual y su transmisión es horizontal, es decir, generalmente sólo hay individuos afectados en una hermandad. En el individuo con FQ las mutaciones se encuentran en ambos alelos del gen CFTR; se les denomina homocigotos cuando éstos portan mutaciones idénticas y heterocigotos compuestos cuando la mutación en cada uno de los alelos es diferente.
Una pareja de portadores (un alelo afectado y uno normal: heterocigotos sanos) en cada embarazo tiene un riesgo de tener hijos afectados de 25%, hijos portadores de 50% e hijos sanos de 25%. En una población, los portadores de una mutación en un gen recesivo siempre son más frecuentes que los afectados (principio de Hardy–Weinberg).13
EL GEN CFTR
En 1989, gracias a la disponibilidad de un gran número de familias con dos o más individuos afectados, se logró la clonación del gen responsable de la FQ (CFTR/ABCC7, OMIM #602421). Este gen se localizó mediante clonación posicional a partir de los marcadores D7515, MET y D758, estrechamente ligados a CFTR.14,15 Los primeros transcritos derivados de este gen se obtuvieron por combinación de mapeo físico e hibridación interespecie; con estos datos se dedujo la secuencia de su producto proteico.16 Así, el gen CFTR se localiza en la banda q31 del cromosoma 7, contiene 250 kb, consta de 27 exones y se transcribe en un mRNA de 6.5 kb (Figura 1).16,17 La identidad del gen se confirmó por la presencia de una deleción de tres pares de bases en el exón 10 (mutación AF508) de los pacientes con FQ que no se observó en individuos normales.18
La región 5' de CFTR se caracteriza por un gran contenido de G–C (aproximadamente 65%) y no presenta caja TATA, de tal manera que su promotor es similar al de los genes con expresión constitutiva, sin embargo, su expresión parece estar altamente regulada y confinada a ciertos tejidos.19,20 Al parecer, los niveles de expresión del gen están modulados por cAMP, PKA, PKC y esteres de forbol. Los patrones de expresión tejido específico de CFTR son similares en las diferentes especies de mamíferos, sugiriendo la presencia de elementos reguladores conservados evolutivamente, aunque se han reportado múltiples sitios de iniciación en el gen que parecen ser especie–tejido–específico.21
CORTE Y EMPALME DEL TRANSCRITO CFTR
En estudios recientes realizados tanto en tejidos de individuos normales como de individuos con FQ se ha logrado identificar, además del mensajero completo de CFTR, varias moléculas de mRNA que han perdido los exones 4, 9 o 12. El transcrito más común es aquel en el que se ha perdido el exón 9 (transcrito 9") que, aunque genera un mRNA con marco de lectura abierto, no se traduce en una proteína funcional y parece no estar conservado entre las especies.22 Este transcrito se encuentra en una proporción que varía de 10–92%, dependiendo de la presencia de un polimorfismo que consiste en tres variantes alélicas de un tracto de politimidinas (5T,7T y 9T) localizado en el brazo aceptor del sitio de corte y empalme del intrón 8; por ejemplo, los homocigotos 5T producen mayores niveles del mRNA sin el exón 9 (89–92%) que los homocigotos 7T (50%) y 9T (8–10%). La eficiencia del corte y empalme del exón 9 parece ser tejido específica, ya que la proporción del transcrito 9– es mayor en células de los vasos deferentes, independientemente del número de timidinas.23 Aunque el significado biológico de estos procesamientos alternativos aún no está claro, en algunas poblaciones se ha observado una asociación entre el alelo 5T y la ausencia bilateral de conductos deferentes (CBAVD), azoospermia obstructiva y bronquiectasias.24
LA PROTEÍNA CFTR
El gen CFTR codifica para una glicoproteína transmembranal de 1,480 aminoácidos y 170 kDa. Esta proteína funciona como un canal de cloro y por su papel fisiológico se denomina proteína reguladora de la conductancia transmembranal de la FQ o CFTR (del inglés "Cystic Fibrosis Transmembrane Conductance Regulator").16 Se expresa en una gran variedad de tejidos especialmente pulmón, páncreas, glándulas sudoríparas, intestino, hígado, mucosa nasal, glándulas salivales y tracto reproductivo. La proteína CFTR presenta una gran homología estructural con la familia de proteínas ABC (cassette de unión a ATP), que son proteínas integrales de membrana que contienen dos segmentos repetidos constituidos por dos dominios transmembranales (TMD1 y TMD2) y dos dominios que interaccionan con ATP (NBD1 y NBD2). A diferencia de las otras proteínas de la familia ABC, CFTR contiene además, un dominio regulador (dominio R) (Figura 1).16,25
La proteína CFTR está anclada a la membrana citoplasmática por los dos TMDs, cada uno organizado en seis segmentos (TM1–TM12) que atraviesan la membrana formando un poro que constituye el canal de cloro. Con base en los efectos que producen algunas mutaciones encontradas en pacientes con FQ, se ha deducido la presencia de varios residuos de aminoácidos en los dominios TMs de CFTR, los cuales son particularmente importantes para la conductancia de iones, como es el caso de los residuos de arginina y asparagina.26
Los dominios NBDs presentan secuencias que se encuentran conservadas en las proteínas que unen e hidrolizan ATP. Interesantemente las estructuras primarias de los dos dominios NBDs sólo presentan 29% de homología entre sus aminoácidos y parece ser que NBD1 y NBD2 contribuyen de manera diferente a la regulación del canal. Se ha propuesto que el NBD1 se fosforila después de una fosforilación parcial del dominio R, lo que permite la apertura del canal, mientras que la fosforilación completa del dominio R, conduce a la fosforilación de NBD2 causando el cierre del canal; de tal manera que el grado de fosforilación del dominio R regula la actividad del canal.25,26
Existen evidencias experimentales que demuestran que la CFTR, además de ser un canal de cloro dependiente de cAMP, puede tener otras funciones, por ejemplo el transporte de bicarbonato y la regulación de otros canales endógenos de cloro y calcio.27,28
MUTACIONES EN EL GEN CFTR
A la fecha el Consorcio Internacional de Fibrosis Quística ha reportado más de 1,300 mutaciones diferentes y alrededor de 200 polimorfismos en el gen CFTR (Figura 2). Su distribución en el gen no parece ser al azar, ya que se ha detectado una alta frecuencia de mutaciones en ciertos exones como el 4, 7, 11, 17b y 20.29

En general la naturaleza de las mutaciones es diversa: cerca de la mitad son de sentido erróneo y el resto son mutaciones sin sentido, pequeñas deleciones o inserciones que alteran el marco de lectura, mutaciones que afectan los sitios de corte y empalme del RNA, grandes deleciones y otras clasificadas como complejas (Figura 2).29 Adicionalmente ciertas mutaciones puntuales en realidad son polimorfismos de un solo nucleótido (SNP's) que potencialmente podrían afectar la expresión de CFTR. Algunos de ellos interfieren con las señales de corte y empalme mientras que otros ocurren en regiones codificadoras del gen; de estos últimos, alrededor de 50% dan lugar a sustituciones de aminoácidos.30,32
Se ha observado una gran heterogeneidad en el espectro de las mutaciones del gen CFTR, particularmente en el Sur de Europa y América Latina (Figura 3). Al parecer este fenómeno está relacionado con la composición étnica de cada región, por ejemplo, la distribución de la mutación más común, la AF508, varía de un máximo del 100% en las Islas Faroe de Dinamarca hasta 24.5% en Turquía.33 En Europa la frecuencia de esta mutación se presenta con un claro gradiente de Noroeste a Sureste. Existen evidencias que sugieren que la mutación AF508 fue introducida al Continente Americano por las migraciones provenientes de Europa, ya que la mutación se ha encontrado asociada al mismo haplotipo que en las poblaciones europeas34 y no se ha detectado en los grupos amerindios estudiados.35 Así, en Estados Unidos y Canadá esta mutación tiene una frecuencia muy elevada (68.6 y 71.4%, respectivamente), similar a la reportada en poblaciones del norte de Europa, mientras que en la población latinomericana la frecuencia de la mutación es más baja, aunque es de notarse que varía entre los diferentes países (Argentina 58.6%, Brasil 47%, México 40.7%, Colombia 35.4%, Venezuela 29.6%, Chile 29.2 y Ecuador 25%).33 Es posible que aunque estas poblaciones están formadas por nativos, blancos, negros y las mezclas de las diferentes razas, estas variaciones se deban a que existen diferencias significativas en las proporciones de los subgrupos de cada país.36

Más aún, estudios realizados en diferentes regiones de México mostraron una frecuencia variable de la mutación AF508 (50% en la región Norte, 34.4% en el Occidente y 40.7% en la región Centro).37–39 Resulta interesante el hecho de que la frecuencia observada en la región Norte es similar a la reportada en España (52.7%),29,33 mientras en el Occidente y Centro, donde 50% de los genes son de origen amerindio, 40% caucásico–hispano y el resto africano,40,41 ésta es más baja. Estas observaciones hacen suponer que durante la época de la Colonia la frecuencia de esta mutación se haya diluido por la mezcla entre poblaciones españolas y amerindias en las regiones Occidente y Centro, mientras que en el Norte esta mezcla ocurrió en menor proporción debido a que las poblaciones nativas mostraron mayor resistencia y llevaban una vida nómada.
Las otras mutaciones en CFTR son poco comunes y aunque algunas son frecuentes en ciertas poblaciones, la mayoría se presentan en menos de 1% de los cromosomas FQ. Inclusive, la segunda mutación más común (G542X), se detecta sólo en 2.4% de los alelos CFTR afectados en todo el mundo, aun cuando en México, Colombia, España y las Islas Canarias, se ha detectado en una proporción más alta (6.1, 6.3, 8 y 25%, respectivamente).39,42,43 De hecho, nuestra población es una de las que ha mostrado mayor número de mutaciones (35 diferentes), incluso siete de ellas se documentaron por primera vez en México: 2055del9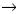 A, 1924del7, W1098C, 846delT, P750L, 4160insGGGG y 297–lGA.39 La complejidad del espectro de las mutaciones observada en nuestros pacientes demuestra la gran diversidad étnica de nuestra población.
A, 1924del7, W1098C, 846delT, P750L, 4160insGGGG y 297–lGA.39 La complejidad del espectro de las mutaciones observada en nuestros pacientes demuestra la gran diversidad étnica de nuestra población.
En poblaciones como la caucásica, donde la mutación ΔF508 es sumamente frecuente, la búsqueda de cinco mutaciones en el gen CFTR (ΔF508, G542X, N1303K, R553X y G551D) logra la detección del 85–90% de los alelos FQ, mientras que en la población mexicana se requiere el análisis de 34 mutaciones para la caracterización de apenas 75% de todos los cromosomas afectados (Cuadro I).39 En poblaciones con una gran diversidad étnica como la nuestra, la detección de las mutaciones en el gen CFTR resulta sumamente compleja y mucho más cara, debido a la gran variedad de mutaciones y la baja frecuencia de la mayoría de ellas.
ANÁLISIS DE HAPLOTIPOS Y MUTACIONES RECURRENTES
Estudios extensos para la identificación de marcadores moleculares dentro y fuera del gen CFTR, han revelado que ciertas mutaciones como la ΔF508, G542X y N1303K se encuentran asociadas con haplotipos únicos, lo que sugiere que cada una de ellas se derivó de un mismo evento mutacional.44 Con base en el análisis de haplotipos se ha sugerido que la mutación AF508 se originó en Europa hace aproximadamente 52,000 años, mientras que la G542X fue introducida a España por los fenicios hace 2,500–3,000 años.45–47 Otras mutaciones como la R117H, H199Y, R334W, R347P, R553X, L558S, 3272–26AG, 3849+10kbC y TR1162X, se encuentran asociadas a más de un haplotipo, por lo que probablemente representen mutaciones recurrentes que surgieron de manera independiente.44
En la población mexicana se estudiaron los haplotipos derivados de dos polimorfismos tipo RFLPs: XV–2C/TaqI (XV) y KM–19/PstI (KM), donde el cambio de un solo nucleótido en la secuencia de DNA, puede crear o eliminar sitios de corte para endonucleasas específicas. Se le denominó alelo 1 cuando el sitio de corte está ausente y alelo 2 cuando está presente. De la combinación de estos polimorfismos se derivaron los siguientes cuatro haplotipos: cuando ninguno de los dos alelos de los polimorfismos XV y KM presentaron el sitio de corte para la enzima de restricción (1/1) se le llamó haplotipo A; cuando el XV no lo presenta pero el KM sí (1/2), se le denominó haplotipo B; cuando ocurre lo contrario y el alelo XV tiene el sitio de corte pero el KM no (1/2), se nombró haplotipo C y cuando tanto XV como KM presentaron el sitio de corte (2/2), se le llamó haplotipo D.34
Los cromosomas con la mutación AF508 se encontraron asociados con el haplotipo B en 97.2%, como se ha reportado en otros países de América del Norte y Europa; sin embargo, la G542X se encontró asociada con este haplotipo sólo en 72.7% de los cromosomas, el resto se asoció con los haplotipos A y D (Cuadro 1). Este hallazgo contrasta con lo reportado en otras poblaciones donde esta mutación se ha encontrado asociada casi exclusivamente con el haplotipo B. Las mutaciones poco frecuentes y las desconocidas, así como los alelos normales, se asociaron con mayor frecuencia a los haplotipos A y C.34 Esta heterogeneidad encontrada en pacientes mexicanos puede ser explicada por:
• Eventos de recombinación
• Mezcla entre poblaciones con distribución de haplotipos diferente.
• La presencia de mutaciones FQ en la población de nativos americanos.
ALELOS COMPLEJOS
En múltiples reportes se describe la coexistencia de dos alteraciones diferentes en el mismo alelo CFTR. Mientras la mayoría de estos alelos complejos representan sólo una asociación de una mutación con una variación benigna o polimorfismo, hay ejemplos en los cuales la presencia de una segunda mutación en el mismo alelo puede modular el efecto de la primera. Por ejemplo, cuando la mutación R553Q se localiza en el mismo alelo que la mutación ΔF508, las manifestaciones clínicas de la enfermedad se atenúan.48 Otro ejemplo es la mutación R117H, que cuando se encuentra en cis con la variante 5T causa una fibrosis quística grave, mientras que cuando se asocia con las variantes 7T o 9T el cuadro clínico es moderado.49
BASES MOLECULARES DE LA DISFUNCIÓN DEL CANAL CFTR
Con la finalidad de entender más acerca de la fisiopatología de la FQ, las mutaciones se han clasificado en cinco clases de acuerdo con la alteración que se genera en la proteína (Figura 4).
Clase I: mutaciones que bloquean la síntesis de la proteína
Cerca de la mitad de las mutaciones en el gen CFTR que pertenecen a este grupo conducen a la formación de proteínas truncadas y no funcionales. Entre ellas se encuentran aquellas que generan codones de terminación (18%), corrimiento del marco de lectura (22%) y las que afectan el proceso de corte y empalme del RNA (8%). Las dos primeras generalmente causan ausencia total de la proteína.22
Clase II: mutaciones que afectan el procesamiento postraduccional
La biosíntesis de la proteína CFTR requiere pasos de maduración complejos, por lo que algunas mutaciones, como la AF508, afectan este proceso y generan péptidos anormales que quedan atrapados en el retículo endoplásmico (RE) donde son degradados por el sistema ubiquitina–proteosoma (vida media de 0.5h).50–51
Clase III: mutaciones que afectan la regulación del canal de cloro
Las mutaciones clasificadas en esta categoría son aquellas que generan proteínas que parecen estar correctamente localizadas en la membrana apical, pero que no funcionan adecuadamente como un canal iónico. Algunas de estas mutaciones afectan los dominios NBDs y el dominio R (Figura 4).52 El grado de alteración puede variar desde una leve reducción de la actividad del canal (mutaciones G551S, G1244E y G1349D) hasta la pérdida grave de su función (mutaciones G551D y S1255P).53
Clase IV: mutaciones que afectan la conductancia del canal de cloro
Estas mutaciones se localizan en regiones del gen que codifican para algunos de los segmentos TMs de la proteína;54 por ejemplo la R117H en la región TM2, la G314E y G314Q en TM5 y la R334W, R347P y R347H en TM6. En estos casos aunque la regulación del canal es adecuada, su conductancia es anormal. Estudios recientes mostraron que algunas mutaciones del dominio R también pertenecen a este grupo.55
Clase V: mutaciones que causan reducción en la síntesis de la proteína
Las mutaciones de esta clase incluyen aquellas localizadas en el promotor, las cuales reducen la transcripción y las mutaciones puntuales que conducen a un procesamiento erróneo del mRNA o a sustituciones de aminoácidos como la P574H y la A455E. Las proteínas mutadas tienen una actividad normal tanto en regulación como en conductancia; sin embargo, su procesamiento postraduccional es ineficiente.22
CORRELACIÓN GENOTIPO–FENOTIPO
Existe una clara variabilidad en la presencia o gravedad de cada una de las manifestaciones clínicas de la FQ y parece ser que esto, en parte, es la consecuencia de la gran variación de las mutaciones en el gen.56 El análisis de la funcionalidad de algunas mutaciones expresadas en ovocitos de Xenopus, reveló que la corriente de cloro generada se encuentra reducida en el siguiente orden: silvestre > mutante con función residual > mutante no funcional,57 lo cual sugiere que existe correlación entre el genotipo y la función del canal de cloro. Diversos estudios han tratado de encontrar las características clínicas que comparten los pacientes con FQ que portan un mismo genotipo (correlación fenotipo–genotipo) y han logrado clasificar a las mutaciones en graves (clase I, II o III) y leves (clases IV o V), donde las primeras ocupan aproximadamente 92% de todas las mutaciones.58,60
De todos los síntomas analizados, sólo la función pancreática correlaciona directamente con el genotipo de los pacientes. En general, los pacientes con IP son homocigotos o heterocigotos compuestos para dos mutaciones graves, mientras que los pacientes con SP son homocigotos o heterocigotos compuestos con al menos una mutación con función residual. Como en todas las enfermedades recesivas, las mutaciones leves son dominantes sobre las graves.58–61
OTROS FENOTIPOS
Una característica clínica frecuente en los pacientes masculinos con FQ es la azoospermia obstructiva y la ausencia bilateral congénita de conductos deferentes (CBAVD). De hecho, la CBAVD aislada puede heredarse como una condición autosómica recesiva, donde se han documentado mutaciones en CFTR en uno o ambos alelos (60–70% y 10% de los casos, respectivamente).62,63 Algunos estudios han mostrado que pacientes con el mismo genotipo presentan CBAVD mientras que otros no, lo que sugiere que otros factores e inclusive otros genes pueden estar involucrados en el desarrollo de esta patología.64 Los pacientes con CBAVD en los que no se han detectado mutaciones en CFTR aparentemente tienen una etiología diferente.12
Otras patologías donde se han encontrado mutaciones en CFTR son la enfermedad pulmonar obstructiva crónica, bronquiectasias difusas, aspergilosis broncopulmonar alérgica, bronquitis crónica por Pseudomona aeruginosa, asma, sinusitis crónica, pólipos nasales, prancreatitis obstructiva crónica, hipertripsinemia neonatal transitoria, azospermia obstructiva idiopática, etc.60,65 Así, las mutaciones en CFTR dan como consecuencia manifestaciones clínicas muy variadas, que pueden ir desde esterilidad primaria como única manifestación clínica hasta el cuadro clásico de FQ, que puede conducir al paciente a la muerte durante los primeros meses de vida.
GENES MODIFICADORES
Con excepción de la función pancreática, el resto de las manifestaciones clínicas de la FQ varía inclusive entre hermanos que comparten el mismo ambiente. Así, la presencia de las diferentes mutaciones en CFTR explica sólo parcialmente la heterogeneidad clínica de la FQ, por lo que se ha propuesto la participación de genes modificadores, que aunque no participan en la etiología de la enfermedad, tienen un gran impacto en la gravedad de las manifestaciones clínicas.66'67 Existen evidencias de que estos genes contienen polimorfismos, principalmente de un solo nucleótido (SNPs), cuyas variantes alélicas son las responsables de la diferencia en la expresividad de la gravedad entre pacientes con un mismo genotipo CFTR.
En esta enfermedad, la gravedad de la afectación pulmonar y la susceptibilidad a infecciones por microorganismos oportunistas puede estar modificada por diversos genes que participan en la respuesta inmune, tales como: HLA Clase II, α1–antitripsina (α1–AT), (α1–antiquimotripsina (α1–ACT), lectina de unión a mañosa (MBL), factor de necrosis tumoral alfa (TNF–α), algunas citosinas y sus receptores, glutation S–transferasa (GST–M1), factor de crecimiento transformante β–1 (TGF–β1), óxido nítrico sintetasa (NOS), receptor β2 adrenérgico (β2AR), entre otros.68,69 Por otro lado, el íleo meconial ocurre sólo en pacientes con IP; sin embargo, se ha documentado que la presencia de esta manifestación se asocia a un locus del cromosoma 19 (19q13.2),70 aunque aún no se han logrado caracterizar genes modificadores en esta región. Otra de las manifestaciones clínicas que parece estar influida por genes modificadores es la afectación hepática, donde se ha observado que ciertos SNPs del gen MBL confieren un mayor riesgo de presentar problemas hepáticos graves.71 Así, la heterogeneidad clínica observada en los pacientes con FQ parece ser la consecuencia de la interacción entre alelos CFTR mutados y un gran número de variantes en otros genes.
DIAGNÓSTICO MOLECULAR
La búsqueda directa de las más de 1,300 mutaciones del gen CFTR o el análisis de su segregación por métodos indirectos utilizando marcadores moleculares ligados al gen, son sin duda la mejor estrategia para el diagnóstico de certeza de las familias con FQ (Figura 5). Se ha documentado que la distribución y frecuencia de las mutaciones varía ampliamente dependiendo del origen étnico de cada población;33,72 de hecho en poblaciones mestizas como la nuestra se observa una gran variación de las mutaciones, incluso algunas de ellas se han encontrado en una sola familia.39 Así, es necesario identificar las mutaciones propias de cada población y establecer estrategias que permitan incrementar la detección de mutaciones en las familias con FQ, realizar el diagnóstico prenatal y presintomático y la detección de portadores heterocigotos para estimar el riesgo de ocurrencia y brindar un asesoramiento genético apropiado. Una de las aportaciones más importantes del diagnóstico molecular es la detección de portadores, ya que clínicamente son indistinguibles de los homocigotos silvestres (sanos) y por los métodos de diagnóstico tradicionales no es posible identificarlos. Así pues, en la FQ el análisis molecular es fundamental para el diagnóstico preciso, el pronóstico, la prevención y el tratamiento oportuno.
Una de las ventajas del diagnóstico molecular, es que puede llevarse a cabo a partir de DNA extraído de una gran variedad de fuentes y realizarse mediante metodología sencilla y cada vez más accesible. Dentro de los métodos más utilizados para el análisis de las mutaciones en CFTR se encuentran: la reacción en cadena de la polimerasa (PCR) y la hibridación específica para la detección de mutaciones conocidas o frecuentes (ASO), el análisis de polimorfismos conformacionales de cadena sencilla (SSCP), cromatografía líquida–desnaturalizante de alta resolución (D–HPLC) y heterodúplex combinados con secuenciación para la caracterización de mutaciones desconocidas y raras.39,73,74 Audrezet, et al., demostraron que el tamizaje de grandes deleciones por PCR múltiple cuantitativo de fragmento corto fluorescente (QMPSF) puede aumentar la detección de alelos FQ.75 Otra alternativa para incrementar la detección de mutaciones son el análisis por Southern blot y los estudios de expresión.
EXPECTATIVAS EN EL MANEJO TERAPÉUTICO
En el tratamiento de la FQ generalmente se prescriben suplementos de enzimas pancreáticas, vitaminas liposolubles, fisioterapia torácica y tratamiento profiláctico con antibióticos. Cuando se presenta íleo meconial u obstrucción del intestino delgado, es necesario realizar una cirugía correctiva. En el caso de pacientes con enfermedad respiratoria terminal y cor–pulmonale el trasplante pulmonar es la mejor alternativa, asimismo se sugiere trasplante hepático en presencia de cirrosis biliar en estado terminal.76,77 Los pacientes con fibrosis quística logran un incremento sustancial en su calidad de vida con el trasplante pulmonar. Sin embargo, dentro de las principales limitantes de este manejo se encuentran la escasez de los donadores, complicaciones tales como síndrome de bronquiolitis obliterante, infecciones o insuficiencia respiratoria, las cuales aunadas al régimen inmunosupresor posterior al trasplante, se asocian con una elevada tasa de mortalidad. Lo anterior y los altos costos de este procedimiento ponen este tipo de tratamiento fuera del alcance de la mayoría de los pacientes, principalmente en países como el nuestro. Aún así, la demanda del trasplante pulmonar va en aumento y actualmente una de las prioridades es la reducción de las complicaciones para mejorar el costo–beneficio en este procedimiento.77
Hasta la fecha no existe un tratamiento definitivo para la FQ; sin embargo, el conocimiento de las bases moleculares de su fisiopatología ha permitido diseñar nuevas estrategias para intentar restablecer la función de la proteína.78 Estas estrategias son:
Administración directa de la proteína CFTR silvestre
En modelos animales se ha logrado corregir el defecto en el transporte iónico introduciendo una proteína CFTR recombinante funcionalmente activa al epitelio nasal de ratones con el gen de fibrosis quística mutado, sin embargo, este efecto es transitorio.79
Inducción de la maduración de la proteína CFTR imitante
Esta estrategia se basa en facilitar la liberación de proteínas mutadas retenidas en el RE, como la AF508, para su posterior procesamiento en el aparato de Golgi (AG) y su ubicación en la membrana apical. Se ha observado que la proteína CFTR de células mutadas ΔF508 pueden formar canales de Cl– con propiedades similares a la silvestre cuando son sometidas a 25 °C in vitro; sin embargo, su aplicación in vivo no ha sido posible ya que estas temperaturas son subfisiológicas.80,81 Aunado a esto, varios estudios han demostrado que solutos orgánicos como el mioinositol pueden reparar la función de la CFTR favoreciendo el procesamiento de la proteína ΔF508, estabilizando su forma glicosilada.82
En este mismo sentido, recientemente Eagan, et al., publicaron que un componente de la especie turmérica, la curcumina, restaura la función de la proteína ΔF508 tanto en células en cultivo como en ratones con FQ.83 Parece ser que su mecanismo de acción es bloquear la entrada de calcio en el retículo endotelial, interfiriendo de esta manera con proteínas chaperonas dependientes de calcio que están involucradas en la degradación de CFTR.84 Por otro lado, trabajos in vitro han mostrado que ciertos aminoglucósidos pueden generar enlaces cruzados con alelos CFTR enmascarando codones de terminación generados por mutaciones sin sentido, permitiendo así, la producción de CFTRs funcionales.85
Inducción de la fosforilación de la CFTR imitante
La fosforilación de la CFTR está altamente controlada por cinasas y fosfatasas. En epitelios normales la fosforilación de los dominios NBDs de la CFTR por la protein cinasa A, regula la apertura y el cierre del canal de Cl–. Se ha demostrado que el transporte de Cl– puede ser estimulado induciendo la fosforilación o inhibiendo la desfosforilación de proteínas CFTRs mutadas.86–88
Estimulación de otros canales de Cl–
Se han identificado otros canales de Cl– en la membrana apical de los epitelios, como los regulados por Ca+ y por volumen, cuyas propiedades y regulación son distintas a las de la proteína CFTR. Se ha observado que cuando los canales de sodio se inhiben con amilorida, los canales de Cl–dependientes de calcio se activan por ATP y UTP extracelulares.89
Terapia génica
La terapia génica consiste en introducir un gen capaz de expresarse dentro de la célula y suplir la función de la proteína alterada y resulta particularmente atractiva para las enfermedades monogénicas que actualmente no tienen opciones de tratamiento definitivo. La fibrosis quística es una de las entidades mendelianas más incluidas en los protocolos de terapia génica.90,91 El blanco inicial para la terapia génica de este padecimiento es el pulmón, ya que éste, además de ser uno de los órganos más comprometidos en el paciente con FQ, es el más accesible. Desde los primeros ensayos de terapia génica se han evaluado numerosos vectores virales y no virales para introducir el cDNA de CFTR a las vías respiratorias.92 Las principales limitantes del uso de los vectores virales en los protocolos de terapia génica han sido: títulos bajos y de transducción del vector, ausencia de regulación de la expresión del transgén, expresión transitoria del gen terapéutico y la respuesta inmune/inflamatoria del hospedero.91 En la mayoría de los ensayos clínicos se ha observado una corrección parcial y transitoria del transporte de cloro92–93 y con la readministración del virus recombinante no se detecta ningún efecto correctivo y la respuesta inflamatoria es muy importante. Por otro lado, las células epiteliales del tracto respiratorio normalmente no son fagocíticas, lo que también limita la utilización de otros vectores no virales como los liposomas, con los que, además, algunos pacientes han presentado efectos adversos en ciertos ensayos.94 Durante los últimos años los investigadores de esta disciplina se han enfocado a buscar o diseñar vectores más eficientes y seguros. Actualmente uno de los vectores con más expectativas para ser utilizados en protocolos clínicos de terapia génica para FQ son los virus–adeno–asociados (AAV), ya que se ha observado que pacientes tratados con estos vectores recombinantes (AAV–CFTR) presentan corrección del transporte de Cl– con poca o ninguna respuesta inflamatoria.95,96 Aún existen barreras que sortear para poder desarrollar estrategias seguras y eficaces para la terapia génica de las enfermedades genéticas, que aunque compleja, resulta muy alentadora.
CONCLUSIONES
A lo largo de esta revisión se discuten los grandes avances en el conocimiento de la FQ, una de las enfermedades monogénicas que ha servido de prototipo para el entendimiento de la fisiopatología de otras enfermedades mendelianas. En los últimos años, los alcances obtenidos del excitante progreso en la investigación de esta patología, incluyendo la identificación del gen y el entendimiento de cómo las mutaciones afectan la función de la proteína CFTR, han sido la piedra angular de la Medicina Molecular, ya que han reforzado nuestro concepto de que el fenotipo de una enfermedad es la suma de componentes clínicos variables que surgen de un genotipo particular, que puede ser modificado por otros factores genéticos secundarios y ambientales. Una de las aportaciones más importantes de estos avances es su aplicación clínica, pues gracias a esto se ha logrado mejorar las estrategias de diagnóstico, prevención y tratamiento de la FQ.
El conocimiento obtenido de la investigación en FQ sienta las bases para el entendimiento molecular y el tratamiento de otras enfermedades monogénicas.
REFERENCIAS
1. Welsh MJ, Ramsey BW, Accuso F, Cutting GR. Cystic fibrosis. In: The metabolic and molecular bases of inherited disease, ed. C Scriver, A Beaudet, W Sly, D Valle, 8° Ed. New York: McGraw–Hill Co.; 2001, pp. 5121–88. [ Links ]
2. Lezana JL, Masa D, Lezana MA. Fibrosis quística en México. Análisis de sus principales aspectos epidemiológicos. Bol Med Hosp Infant Mex 1994; 5: 305–10. [ Links ]
3. Davis PB. Cystic Fibrosis. Pediatr Rev 2001; 2: 257–64. [ Links ]
4. Munger B, Brusilow S, Cooke R. An electron microscopic study of eccrine sweat glands in patients with cystic fibrosis of the pancreas. J Pediatr 1961; 59: 497–11. [ Links ]
5. Tomashefski JF Jr, Bruce M, Stern RC, Dearborn DG, Dahms B. The pathology of pulmonary air cysts in cystic fibrosis. Relation to radiologic findings and history of pneumothorax. Hum Pathol 1985; 16: 253–61. [ Links ]
6. Staab D. Cystic fibrosis: Therapeutic challenge in cystic fibrosis children. Eur J Endocrinol 2004; 151: 577–80. [ Links ]
7. Mischler EH, Chesney PJ, Chesney RW, Mazes RB. Demineralization in cystic fibrosis. Am J Dis Child 1979; 133: 632–5. [ Links ]
8. Conwal SP. Vitamin K in cystic fibrosis. J R Soc Med 2004; 97: 48–51. [ Links ]
9. Anderson D. Cystic fibrosis of the pancreas and its relationship to celiac disease: Clinical and pathologic study. Am J Dis Child 1938; 56: 344–99. [ Links ]
10. Stern RC, Stevens DP, Boat TF, Doershuk CF, Izant RF, Matthews LW. Symptomatic hepatic disease in cystic fibrosis: incidence, course, and outcome of portal systemic shunting. Gastroenterology 1976; 70: 645–9. [ Links ]
11. Oliveira MC, Reis FJ, Monteiro AP, Penna FJ. Effect of meconium ileus on the clinical prognosis of patients with cystic fibrosis. Braz J Med Biol Res 2002; 35: 31–8. [ Links ]
12. Jarzabet K, Zbucka M, Pepinski W, Szamatowicz J, Domitrz J, Janica J, Wotezynski S, Szamatowicz M. Cystic fibrosis as a cause of infertility. Reprod Biol 2004; 2: 119–29. [ Links ]
13. Orozco L, Alcántara MA, González A. Diagnóstico molecular de las enfermedades hereditarias. La frontera: Genética molecular de la enfermedad. Ed JP Luna, E. Orozco; México: Instituto Politécnico Nacional 2004; 2: 46–74. [ Links ]
14. Tsui L, Buchwald M, Barker D, Braman JC, Knowlton R, Schumm JW, et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science 1985; 29: 1054–7. [ Links ]
15. Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245: 1059–65. [ Links ]
16. Riordan JR, Alon N, Grzelczak Z, Dubel S, Sun S. The CF gene product as a member of a membrane transporter (TM6–NBF) superfamily. Adv Exp Med Biol 1991; 290: 19–29. [ Links ]
17. Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 1991; 10: 214–28. [ Links ]
18. Kerem BS, Rommens J, Buchanan J, Durie P, Corey ML, Levison H, et al. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989; 245: 1074–80. [ Links ]
19. Chou JL, Rozmahel R, Tsui LC. Characterization of the promoter region of the cystic fibrosis transmembrane conductance regulator gene. J Biol Chem 1991; 266: 24271–476. [ Links ]
20. Yoshimura K, Nakamura H, Trapnell BC, Chu Cs, Daleman W, Pavirani A. The cystic fibrosis gene has a "housekeeping"–type promoter and is expressed a low levels in cell of epithelial origin. J Biol Chem 1991; 226: 9140–4. [ Links ]
21. White NL, Higgins CF, Trezise AEO. Tissue–specific in vivo transcription start site of the human and murine cystic fibrosis genes. Hum Mol Genet 1998; 7: 363–9. [ Links ]
22. Zielenski J, Tsui LC. Cystic fibrosis: Genotypic and phenotypic variations. Annu Rev Genet 1995; 29: 777–807. [ Links ]
23. Teng H, Jorissen M, Poppel H, Legius E, Cassiman JJ, Cuppens H. Increased proportion of exon 9 alternatively spliced CFTR transcripts in vas deferents compared with nasal epithelial cells. Hum Mol Genet 1997; 6: 85–90. [ Links ]
24. Chillón M, Casals T, Mercier B, Bassas L, Lissens W. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferents. N Engl J Med 1995; 332: 1475–80. [ Links ]
25. Riordan JR. Assembly of functional CFTR chloride channels. Annu Rev Physiol 2005; 67: 701–18. [ Links ]
26. Akabas MH. Cystic fibrosis transmembrane conductance regulator. Structural and function of an epithelial chloride channel. J Biol Chem 2000; 275: 3729–32. [ Links ]
27. Choy JY, Muallem D, Kiselyov K, Lee MG, Thomas PJ, Muallen S. Aberrant CFTR– dependent HC03–transport in mutation associated with cystic fibrosis. Nature 2001; 410: 94–7. [ Links ]
28. Vankeerberghen A, Cuppens H, Cassiman JJ. The cystic fibrosis transmembrane conductance regulator: and intriguing protein with pleiotropic functions. J Cyst Fibros 2002; 1: 13–29. [ Links ]
29. Cystic Fibrosis Genetic Analysis Consortium. http//www.genet.sickkids.on.ca [ Links ]
30. Rowntree R, Harris A. DNA polymorphisms in potential regulatory elements of the CFTR gene alter transcription factor binding. Hum Genet 2002; 111: 66–74. [ Links ]
31. Tzetis M, Efthymiadou A, Doudounakis S, Kanavakis E. Qualitative and quantitative analysis of mRNA associated with four putative splicing mutations (621+3 AG, 2751+2TA, 296+lGC, 1717–9T C–D565G) and one nonsense mutation (E822X) in the CFTR gene. Hum Genet 2001; 109: 592–601. [ Links ]
32. Cartegni L, Chew SL, Kriner AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002; 3: 285–98. [ Links ]
33. Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: A world wide analysis of the CFTR mutations–correlation with incidence data and application to screening. Hum Mutat 2002; 19: 575–606. [ Links ]
34. Orozco L, González L, Chavez M, Velazquez R, Lezana JL, Saldaña Y, et al. XV–2c/KM–19 haplotype analysis of cystic fibrosis mutations in Mexican patients. Am J Med Genet 2001; 102: 277–81. [ Links ]
35. Grebe TA, Doane WW, Richter SF, Clericuzio C, Norman RA, Seltzer WK, et al. Mutation analysis of the cystic fibrosis transmembrane regulator gene in Native American populations of the southwest. Am J Hum Genet 1992; 51: 736–40. [ Links ]
36. Arzimanoglou II, Tuchman A, Li Z, Gilbert F, Denning C, Valverde K, et al. Cystic fibrosis carrier screening in Hispanics. Am J Hum Genet 1995; 56: 544–7. [ Links ]
37. Villalobos C, Rojas A, Villarreal E, Cantú JM, Sanchez FJ, Saiki RK, et al. Analysis of 16 cystic fibrosis mutations in Mexican patients. Am J Med Genet 1997; 69: 380–2. [ Links ]
38. Flores S, Gallegos M, Moran M, Sánchez J. Detection of AF508 mutation in cystic fibrosis patients from northwestern Mexico. Ann Genet 1997; 40: 189–91. [ Links ]
39. Orozco L, Velazquez R, Zielenski J, Tsui LC, Chavez M, Lezana JL, et al. Spectrum of CFTR mutations in Mexican cystic fibrosis patients: identification of five novel mutations (W1098C, 846delT, P750L, 4160insGGGG and 297–1G A). Hum Genet 2000; 106: 360–5. [ Links ]
40. Lisker R, Pérez–Briceño R, Granados J. Gene frequencies and admixture estimates in a Mexico City population. Am J Phys Anthropol 1986; 71: 203–7. [ Links ]
41. Lisker R, Ramírez E, Pérez–Briceño R, Granados J, Babinsky V. Gene frequencies and admixture estimates in four Mexican urban centers. Hum Biol 1990; 62: 791–801. [ Links ]
42. Restrepo MC, Pineda L, Rojas–Martínez A, Gutiérrez CA, Morales A, Gómez Y, et al. CFTR mutations in three Latin American countries. Am J Med Genet 2000; 91: 4, 277–9. [ Links ]
43. Casals T, Nunes V, Palacios A, Giménez J, Gaona A, Ibanez N, et al. Cystic fibrosis in Spain: High frequency of mutation G542X in the Mediterranean coastal area. Hum Genet 1993; 91: 66–70. [ Links ]
44. Morral N, Dotk T, Dziadek V, Llevadot R, Ferec C. Patterns of haplotypes for 92 cystic fibrosis mutations: variability, association and recurrence. Am J Hum Genet 1994; 55: 918. [ Links ]
45. Niu T, Qin ZS, Xu X, Liu JS. Bayesian haplotype inference for multiple linked single–nucleotide polymorphisms. Am J Hum Genet 2002; 70: 157–69. [ Links ]
46. Morral N, Bertranpetit J, Estivill X, Nunes V, Casals T. The origin of the major cystic fibrosis mutation (delta–F508) in European populations. Nat Genet 1994; 7: 169–75. [ Links ]
47. Loirat F, Hazout S, Lucotte G. G542X as a probable Phoenician cystic fibrosis mutation. Hum Biol 1997; 3: 419–25. [ Links ]
48. Dork T, Wulbrand U, Richter T, Neumann T, Wolfes H. Cystic fibrosis with three mutations in the cystic fibrosis transmembrane conductance regulator gene. Hum Genet 1991; 87: 441–6. [ Links ]
49. Kiesewetter S, Macek M Jr, Davis C, Curristin SM, Chu CS, Graham C, et al. A mutation in CFTR produces different phenotypes depending on chromosomal background. Nat Genet 1993; 5: 274–8. [ Links ]
50. Cheng SH, Gregory RJ, Marshall J, Paul S, Suoza DW. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990; 63: 827–34. [ Links ]
51. Amaral MD. CFTR and chaperone: processing and degradation. J Mol Neurosci 2004; 23: 41–8. [ Links ]
52. Hiestad DM, Sorcher EJ, Huang Z, Wang Y, Haley BE. Use of 2–N3ATP to identify the site of ATP interaction on nucleotide binding domain–2 from cystic fibrosis transmembrane conductance regulator. Pediatr Pulmonol 1994; 10: 42. [ Links ]
53. Carson MR, Travis SM, Welsh MJ. The two nucleotide–binding domains of the cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J Biol Chem 1995; 270: 1711–17. [ Links ]
54. Mansoura MK, Smith SS, Choi AD, Richards NW, Strong TV, Drum ML, et al. Cystic fibrosis tansmembrane conductance regulator (CFTR) anion binding has a probe of the pore. Biophys J 1998; 74: 1320–32. [ Links ]
55. Vankeerberghen A, Wei L, Jaspers M, Cassiman JJ, Nilius B and Cuppens H. Characterization of 19 disease–associated missense mutations in the regulatory domain of the cystic fibrosis transmembrane conductance regulator. Hum Mol Genet 1998; 7: 1761–9. [ Links ]
56. Groman JD, Meyer ME, Wilmott RW, Zeitlin TL, Cutting GR. Variant cystic fibrosis phenotypes in the absence of CFTR. N EnglJ Med 2002; 347: 401–7. [ Links ]
57. Sheppard ND, Ostedgaard SL. Understanding how cystic fibrosis mutations caused a loss of chloride channel function. Mol Med Today 1996; 290–7. [ Links ]
58. Kerem B, Corey M, Kerem BS, Rommens J, Markiewicz D. The relation between genotype and phenotype in cystic fibrosis—analysis of the most common mutation (delta F508). N Engl J Med 1990; 323: 1517–22. [ Links ]
59. Durno C, Corey M, Zielenski J, Tullís E, Tsui LC, Durie P. Genotype and phenotype correlation in patients with cystic fibrosis and pancreatitis. Gastroenterology 2002; 123: 1857–64. [ Links ]
60. Doull I. Recent advances in cystic fibrosis. Arch Dis Child 2001; 85: 62–6. [ Links ]
61. Noone PG, Knowles MR. "CFTR–opathies": disease phenotypes associated with cystic fibrosis transmembrane regulator gene mutations. Respir Res 2001; 2: 328–32. [ Links ]
62. Schellen TM, van Straaten A. Autosomal recessive hereditary congenital aplasia of the vasa deferentia in four siblings. Fertil Steril 1980; 35: 401–4. [ Links ]
63. Rowntree RK and Harris A. The phenotypic consequences of CFTR mutations. Ann Hum Genet 2003; 67: 471–85. [ Links ]
64. Rave–Harel N, Madgar I, Goshen R, Nissim–Rafinia N, Ziadni A, Rahat A, et al. CFTR haplotype analysis reveals genetic heterogeneity in the etiology of congenital bilateral aplasia of the vas deferens. Am J Hum Genet 1995; 56: 1359–66. [ Links ]
65. Ninis VN, Kylync MO, Kandemir M, Dadly E, Tolun A. High frequency of T9 and CFTR mutations in children with idiopathic bronchiectasis. J Med Genet 2003; 40: 530–5. [ Links ]
66. Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration 2000; 67: 117–33. [ Links ]
67. Sontag MK, Accurso FJ. Gene modifiers in pediatrics: application to cystic fibrosis. Adv Pediatr 2004; 51: 5–36. [ Links ]
68. Drumm ML. Modifier genes and variation in cystic fibrosis. Respir Res 2001; 2: 125–8. [ Links ]
69. Acton JD, Wilmott RW. Phenotype of CF and the effects of possible modifier genes. Pediatr Res Rev 2001; 2: 332–9. [ Links ]
70. Zielenski J, Corey M, Rozmahel R, Markiewics D, Aznares I, Casals T, et al. Detection of a cystic fibrosis modifier locus for meconium ileus on human chromosome 19q13. Nat Genet 1999; 22: 128–9. [ Links ]
71. Salvatore F, Scudiero O, Castaldo G. Genotype–Phenotype Correlation in cystic fibrosis: the role of modifier genes. Am J Med Genet 2002; 111: 88–95. [ Links ]
72. Mateu E, Calafell F, Lao O, Bonne–Tamir B, Kidd JR. Worldwide genetic analysis of the CFTR region. Am J Hum Genet 2001; 68: 103–17. [ Links ]
73. Ravnick–Glavac M, Atkinson A, Glavac D, Dean M. DHPLC screening of cystic fibrosis gene mutations. Hum Mutat 2002; 19: 374–83. [ Links ]
74. D'Apice MR, Gambardella S, Bengala M, Russo S, Nardone AM, Lucidi V, et al. Molecular analysis using DHPLC of cystic fibrosis: increase of the mutation detection rate among the affected population in Central Italy. BMC Med Genet 2004; 5: 1–7. [ Links ]
75. Audrezet MP, Chen JM, Raguenes O, Chuzhanova N, Giteau K, Le Marechal C, et al. Genomic rearrangements in the CFTR gene: extensive allelic heterogeneity and diverse mutational mechanisms. Hum Mutat 2004; 23: 343–57. [ Links ]
76. Fridell JA, Bond GJ, Mazariegos GV, Orenstein DM, Jain A, Sindhi R, et al. Liver transplantation in children with cystic fibrosis: a long–term longitudinal review of a single center's experience. J Pediatr Surg 2003; 38: 1552–6. [ Links ]
77. Meyers BF, De la Morena M, Sweet SC, Trulock EP, Guthrie TJ, Mendeloff EN, et al. Primary graft dysfunction and other selected complications of lung transplantation: A single–center experience of 983 patients. J Thorac Cardiovasc Surg 2005; 129: 1421–9. [ Links ]
78. Guggino WB, Banks–Schlegel SP. Macromolecular interactions and ion transport in cystic fibrosis. Am J Respir Crit Care Med 2004; 170: 815–20. [ Links ]
79. Ramjeesingh M. Treatment of the nasal epithelium of CF mice with liposomes containing purified CFTR protein. Pediatr Pulmonol 1995; 12: S10.7. [ Links ]
80. Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature–sensitive. Nature 1992; 358: 761–4. [ Links ]
81. Tonghui M, Ventrivel M, Yang H, Pedemonte N, Zegarra–Morán O, Galietta LJ, Verkman AS. High–affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high–throughput screening. J Biol Chem 2002; 277: 37235–41. [ Links ]
82. Zhang XM, Wang XT, Yue H, Leung SW, Thibodeau PH, Thomas PJ, et al. Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J Biol Chem 2003; 278: 51232–42. [ Links ]
83. Egan ME, Pearson M, Weiner SA, Rajendran V, Rubin D, Glockner–Pagel J, et al. Curcumin, a major constituent of turmeric correct cystic fibrosis defect. Science 2004; 304: 600–2. [ Links ]
84. Davis PB, Drumm ML. Some like it hot: curcumin and CFTR. Trends Mol Med 2004; 10: 473–5. [ Links ]
85. Kerem E. Pharmacologic therapy for stop mutations: how much CFTR activity is enough? Curr Opin Pulm Med 2004; 10: 547–52. [ Links ]
86. Drumm ML, Kelley TJ. Inhibition of specific phosphodiesterases in CF airway epithelial cells activates mutant CFTRs. Pediatr Pulmonol 1995; 12(Supl): 150–1. [ Links ]
87. Howell LD, Borchardt R, Kole J, Kaz AM, Randak C, Cohn JA. Protein kinase A regulates ATP hydrolysis and dimerization by a CFTR (cystic fibrosis transmembrane conductance regulator) domain. Biochem J 2004; 378: 151–9. [ Links ]
88. Csanady L, Seto–Young D, Chan KW, Cenciarelli C, Angel BB, Qin J, et al. Preferential phosphorylation of R–domine serine 768 dampens activation of CFTR channels by PKA. J Gen Physiol 2005; 125: 171–86. [ Links ]
89. Mason SJ, Paradiso AM, Boucher RC. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. Br J Pharmacol 1991; 103: 1649–56. [ Links ]
90. Griesenbach U, Geddes DM, Alton EW. Gene therapy for cystic fibrosis: an example for lung gene therapy. Gene Ther 2004; 11: S43–S50. [ Links ]
91. Driskell RA, Engelhardt JF. Current status of gene therapy for inherited lung diseases. Annu Rev Physiol 2003; 65: 585–612. [ Links ]
92. Zabner J, Couture LA, Gregory RJ, Graham SM, Smith AE, Welsh MJ. Adenovirus–mediated gene transfer transiently corrects the chloride transport defect in nasal epithelial of patients with cystic fibrosis. Cell 1993; 75: 207–16. [ Links ]
93. Zabner J, Ramsey BW, Meeker DP, Aitken ML, Balfour RP. Repeat administration of an adenovirus vector encoding cystic fibrosis transmembrane conductance regulator to the nasal epithelium of patients with cystic fibrosis. J Clin Invest 1996; 97: 1504–11. [ Links ]
94. Ruiz FE, Clancy JP, Perricone MA, Bebok Z, Hong JS. A clinical inflammatory syndrome attributable to aerosolized lipid–DNA administration in cystic fibrosis. Hum Gene Ther 2001; 12: 751–61. [ Links ]
95. Aitken ML, Moss RB, Walts DA, Dovey ME, Tonelli MR. A phase I study of aerosolized administration of tg AAVCF to cystic fibrosis subjects with mild lung disease. Hum Gen Ther 2001; 12: 1907–16. [ Links ]
96. Moss RB, Rodman D, Spencer LT, Aitken ML, Zeitlin PL, Waltz D, et al. Repeated adeno–associated virus serotype 2 aerosol–mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis multicenter, double–blind, placebo–controlled trial. Chest 2004; 125: 509–21. [ Links ]

