










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de investigación clínica
versión On-line ISSN 2564-8896versión impresa ISSN 0034-8376
Rev. invest. clín. vol.58 no.2 Ciudad de México mar./abr. 2006
Artículo original
Patrón clínico y hematológico de pacientes con drepanocitosis o diagnóstico presuntivo de talasemia
Hematological and clinical profile in sickle cell or thalassemic patients
Daniella Vilachá,* Raquel Salazar*
* Laboratorio de Proteínas e Inmunotoxicidad. Departamento de Bioanálisis, Escuela de Ciencias, Núcleo de Sucre, Universidad de Oriente, Cumaná, Estado Sucre, Venezuela.
Reimpresos:
Raquel Salazar Lugo
Lab. Proteínas e Inmunotoxicidad, Posgrado de Biología Aplicada.
Cerro del Medio, Núcleo de Sucre, Universidad de Oriente.
Apartado Postal 106.
Tel. y fax. 0293 4317020.
Correo electrónico: raquelugove@yahoo.com
Recibido el 14 de abril de 2004.
Aceptado el 1 de diciembre de 2005.
ABSTRACT
Clinical and hematological characteristics of 14 patients with sickle cell anemia; one heterozygous AS, and 7, with diagnostic of microcytic hypochromic anemia were analyzed. Hemoglobin phenotypes were identified by electrophoresis, fetal hemoglobin was quantificated for alkaline denaturation and the HbA2 for ionic exchange chromatography; –α3,7–thalassemia was detected by mutation identification using polymerase chain reaction (PCR). SS phenotype was confirmed in 10 patients, two were SSF , one was SSFA2, and one was ASF (HbF – 2%). The patient diagnosed as AS was SSF (HbF = 21%). AD–patients presented a moderate clinical course of the illness. Five microcytic hypochromic anemia patients were HbAA, one was HbAAA2 and another HbAAF; those patients present a high hematological and clinical variation, β–thalassemia was 19%. –α3,7 –thalassemia was not detected. Infection was most frequent clinical manifestation (respiratory tract infection and intestinal parasitism). These results shows that –α3,7 –thalassemia are not modulator genetic factors of clinical and hematological manifestations of patients with microcytic hypochromic anemia and sickle cell anemia. We suggest that environmental factors such as respiratory tract infection and intestinal parasitism may be affect the course of illness.
, one was SSFA2, and one was ASF (HbF – 2%). The patient diagnosed as AS was SSF (HbF = 21%). AD–patients presented a moderate clinical course of the illness. Five microcytic hypochromic anemia patients were HbAA, one was HbAAA2 and another HbAAF; those patients present a high hematological and clinical variation, β–thalassemia was 19%. –α3,7 –thalassemia was not detected. Infection was most frequent clinical manifestation (respiratory tract infection and intestinal parasitism). These results shows that –α3,7 –thalassemia are not modulator genetic factors of clinical and hematological manifestations of patients with microcytic hypochromic anemia and sickle cell anemia. We suggest that environmental factors such as respiratory tract infection and intestinal parasitism may be affect the course of illness.
Key words. Sickle cell anemia. Venezuela. Thalassemia. Sucre state.
RESUMEN
Se analizaron las características clínicas y hematológicas de 14 pacientes con diagnóstico clínico de anemia drepanocítica (AD), un heterocigoto AS con manifestaciones clínicas, y siete pacientes con diagnóstico de anemia microcítica hipocrómica resistente a tratamiento con hierro y ácido fólico. Los fenotipos hemoglobínicos fueron determinados mediante electroforesis, la cuantificación de hemoglobina fetal se realizó por desnaturalización alcalina y la hemoglobina A2 por cromatografía de intercambio iónico. La detección de –α3,7 talasemia se realizó mediante la técnica de reacción en cadena de polimerasa (PCR). Se confirmó el fenotipo SS para 10 pacientes; de los cuatro restantes, dos fueron SSF, uno SSFA2, y uno fue heterocigoto ASF (HbF = 2%). El paciente diagnosticado como heterocigoto AS resultó ser SSF (HbF = 21%). Los pacientes con AD presentaron un curso clínico moderado de la enfermedad. De los siete pacientes con anemia microcítica hipocrómica, cinco fueron HbAA, uno fue HbAAA2 y otro HbAAF; todos presentaron una alta variación hematológica y clínica. Se detectó la presencia de β–talasemia en 19% de los pacientes. No se detectó la presencia de –α3,7 –talasemia. La manifestación clínica más frecuente fue la infección (respiratoria o parasitismo intestinal). De acuerdo con estos resultados, en estos pacientes se descarta la presencia de –α3,7 –talasemia, como atenuante de las manifestaciones clínicas de la anemia drepanocítica y como factor modulador de la variabilidad clínica observada en los pacientes con anemia microcítica–hipocrómica; se sugiere que factores ambientales tales como parasitosis intestinales y enfermedades respiratorias pueden afectar el curso de la enfermedad.
Palabras clave. Drepanocitosis. Venezuela. Talasemia. Estado Sucre.
INTRODUCCIÓN
La razón más frecuente de consulta hematológica es la presencia de anemia; muchas de estas anemias son producidas por factores genéticos y entre éstos los más comunes son la presencia de talasemias (alfa y beta) y de variantes hemoglobínicas, en particular, la hemoglobina S. Entre las α–talasemias, el tipo más común se produce por entrecruzamiento entre dos regiones diferentes del bloque de genes alfa generando la pérdida de un fragmento de 3,7 kb. La mutación –α3 talasemia, se encuentra presente en todos los grupos étnicos en áreas tropicales y subtropicales del mundo.1,2
La anemia drepanocítica (AD), producida por la presencia de dos genes mutantes S, cursa con ciertas manifestaciones clínicas, que en muchos casos conllevan a la muerte prematura en la mayoría de los pacientes. Los sujetos portadores del rasgo drepanocítico o heterocigotos (HbAS), usualmente, son asintomáticos; sin embargo, se han reportado casos con severas manifestaciones clínicas similares al estado homocigoto.3
En la intensidad del cuadro clínico de la anemia drepanocítica influyen factores ambientales y genéticos. Diversos estudios demuestran que las manifestaciones clínicas en estado homocigoto, varían entre individuos y de acuerdo con las diferentes localizaciones geográficas de las poblaciones estudiadas, asumiendo que la expresión clínica y hematológica de la anemia drepanocítica puede ser modificada por otros determinantes genéticos llamados fenómenos epigenéticos. Se sugiere que la presencia concomitante de la α–talasemia con la anemia drepanocítica o niveles elevados de la HbF, benefician el estado clínico de la enfermedad.4,5
Existe una alta incidencia del gen drepanocítico en algunas poblaciones del Estado Sucre; incluso se ha detectado la presencia de alfa talasemia en pacientes drepanocíticos en otras regiones de Venezuela.2,6 Sin embargo, son escasos los trabajos reportados sobre el patrón de morbilidad de estos pacientes en nuestro país. En consecuencia, se planteó la necesidad de evaluar las características clínicas y hematológicas, en pacientes con diagnóstico clínico de drepanocitosis o presuntivo de talasemia y detectar la presencia de la delección 3,7 del gen α–globina.
METODOLOGÍA
Se incluyeron en el estudio 21 pacientes, 14 con diagnóstico clínico de anemia drepanocítica (AD), un portador o tara drepanocítica y siete con anemia microcítica hipocrómica resistente al tratamiento con hierro y ácido fólico (presuntivo de α–talasemia), que acudieron a consulta hematológica del Banco de Sangre del Hospital Universitario "Antonio Patricio de Alcalá" de la Ciudad de Cumaná (SAHUAPA), Estado Sucre, Venezuela. Todos los pacientes se presentaron en condiciones estables.
Para este estudio se contó con el permiso y la aprobación por escrito del paciente o representante, así como con la colaboración del médico tratante; siguiendo las normas de ética establecidas para trabajos de investigación en humanos, por la Organización Mundial de la Salud (OMS) y la Declaración de Helsinki ratificada por la 29th World Medical Assembly, Tokio 1975.
Se realizó análisis hematológico de hemoglobina (Hb), hematócrito (Hcto), recuento eritrocitario y leucocitario, volumen corpuscular medio (VCM), concentración de hemoglobina corpuscular media (CHCM) e índice de distribución de la amplitud eritrocitaria (RDW); con la ayuda del equipo automatizado Cell–Dyn 1600. Se prepararon frotis de sangre anticoagulada y se colorearon con Giemsa para el recuento diferencial leucocitario. Conjuntamente, se desarrolló un estudio retrospectivo, basado en los expedientes clínicos de los pacientes.
Determinación del fenotipo hemoglobínico
El fenotipo hemoglobínico fue determinado por electroforesis a pH 8,6 en acetato de celulosa,7 utilizando un equipo de Helena Laboratories. Para separar HbS de la HbD se realizó una electroforesis empleando bacto agar (Difco Laboratories) al 1.5% (p/ v) en buffer citrato a pH 6,2 (C6HNa307.2H20 500 mmol/L). La cuantificación de hemoglobina fetal (HbF) se realizó por el método de desnaturalización alcalina modificado.8 La cuantificación de HbA2 se realizó por cromatografía en columna de intercambio iónico.9,10
Detección de α–talasemia3,7
La determinación de α–talasemia fue realizada en el Laboratorio de Hemoglobinas Anormales "Dr. Tulio Arends" del Hospital Clínico Universitario de Caracas. La separación del material genómico fue realizada a partir de muestras congeladas de sangre venosa.11 La extracción del DNA se realizó de acuerdo con Jeampiere12 modificado. El DNA fue cuantificado mediante espectrofotometría, en un equipo GeneQuant (Bio Rad).
La detección de la delección 3,7 de α–talasemia mediante la técnica de PCR, fue hecha según protocolo de Baysal y Huisman.13 Los productos amplificados fueron corridos en electroforesis en gel de agarosa al 2% y buffer TBE IX, por una hora 30 minutos a 100 V. Los fragmentos de restricción fueron visualizados con bromuro de etidio en un transiluminador ultravioleta (UV).
Análisis estadístico
A los pacientes con anemia drepanocítica y anemia microcítica, se les determinaron las correlaciones entre las variables hematológicas y los parámetros clínicos, mediante el índice de correlación producto o momento r de Pearson, considerando los cuatro hallazgos clínicos más importantes en ambos grupos. Se tomaron en cuenta valores de r mayores o iguales a 0.50 para definir una relación confiable. Finalmente, se aplicó la prueba t–Student, para establecer la significancia de la relación. Todo el análisis estadístico de los datos se realizó a un nivel de confiabilidad del 95%, y con la ayuda del programa Microsoft Excel 2000 para Windows.
RESULTADOS
El análisis electroforético realizado a los 14 individuos con diagnóstico clínico de AD, reveló que 13 resultaron homocigotos SS y uno fue heterocigoto ASF (HbF: 2%). De los homocigotos SS, dos fueron SSF; uno fue SSFA2; mientras que el paciente con diagnóstico de portador o tara drepanocítica, se le comprobó el fenotipo homocigoto SSF (HbF: 21%). Por otro lado, de los siete individuos con diagnóstico de anemia microcítica–hipocrómica, cinco fueron HbAA, uno fue HbAAA2 y otro HbAAF Ninguno de los productos de amplificación para el gen mutante reveló la delección 3,7 del gen alfaglobina, ni en los pacientes drepanocíticos ni en los diagnosticados con anemia microcítica–hipocrómica.
Se logró determinar el hallazgo de al menos cuatro (19%) individuos con β–talasemia. Dentro de éstos se encuentran dos de los pacientes con diagnóstico clínico de AD: el que presentó el fenotipo βS–homocigoto compuesto, fetal aumentada y manifestaciones clínicas moderadas (HbSSFA2); y el otro, β talasémico con fenotipo βS–heterocigoto compuesto, fetal aumentada y manifestaciones clínicas moderadas (HbASF).
De los pacientes con anemia microcítica hipocrómica; uno fue β–talasémico con fenotipo de hemoglobina normal, fetal aumentada y curso clínico complicado (HbAAFT), y otro resultó β–talasémico con fenotipo de hemoglobina normal y curso clínico benigno (HbAAF). Ambos presentaron valores de Hb A2 entre 2.5–3%.
Los individuos SS cursaron con niveles moderados de anemia (Hb 8.6 ±1.1 g/dL), índices hematimétricos promedios normales, eosinofilia e índice RDW elevado; no se halló diferencia significativa (p > 0.05) entre niños y adultos. Los pacientes AA mostraron anemia microcítica hipocrómica (Hb 9.5 ± 2.3 g/dL), con valores significativamente mayores de GR (p < 0.001) y menores de VCM (p < 0.01) e índice RDW (p < 0.001), con respecto a los pacientes SS (Cuadro 1). Se observó mayor variación en los parámetros hematológicos eritrocitarios (Hb, Hcto, GR e índices hematimétricos) en los individuos AA (coeficiente de variación 1 18%), que en los pacientes SS (coeficiente de variación 19%).
El 100% de los pacientes con AD (ocho niños y seis adultos) fue hospitalizado al menos una vez durante su vida y el motivo más frecuente de admisión fue el síndrome anémico, acompañado de dolor y fiebre persistente. En los niños, la edad del primer ingreso resultó significativamente menor que en los adultos, y fueron hospitalizados con mayor frecuencia que los adultos (Cuadro 2).
Se observó que niveles bajos de hemoglobina y hematócrito basal presentaron correlación (r = –0.53 y r = –0.56) con un mayor número de crisis drepanocíticas en los niños; mientras que en los adultos, valores altos de VCM se correlacionaron con un mayor número de ingresos (r = 0.69) y de crisis drepanocíticas (r = 0.96).
El 71% de los pacientes AA (cuatro niños y una mujer) fue alguna vez hospitalizado. No se hallaron diferencias significativas (p > 0.05) con los pacientes SS en cuanto al número de ingresos anuales, edad de la primera hospitalización o la edad diagnóstico de la anemia (Cuadro 3).

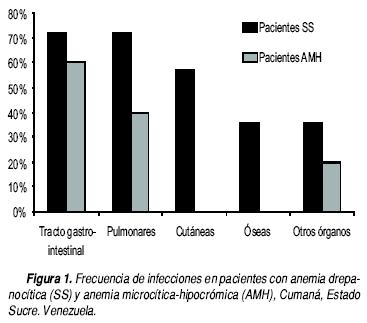
Al igual que en los pacientes SS, las infecciones gastrointestinales, respiratorias y las transfusiones, fueron los hallazgos clínicos más frecuentes (Cuadro 4, Figura 1). El diagnóstico clínico más común fue anemia crónica persistente (57%). En la revisión de la historia clínica de estos pacientes, no se reportaron diagnósticos de β talasemia. No se encontró correlación estadística entre la clínica y la hematología de los pacientes AA.
DISCUSIÓN
En general, la historia clínica de los pacientes con AD, reveló un curso moderado de la enfermedad, es decir, sin la presencia de manifestaciones severas comunes en estos pacientes tales como síndrome agudo de pecho, cardiomegalia, deformaciones óseas, entre otras. Los niños fueron hospitalizados con mayor frecuencia que los adultos. Aunque las transfusiones fueron comunes, el número de las mismas fue menor a las referidas por otros estudios.14,15
Los valores de Hb y Hcto de los niños SS, mostraron correlación con el número de hospitalizaciones. Otras investigaciones evidencian que niveles elevados de hemoglobina total, son un factor de riesgo para las crisis dolorosas y necrosis de la cabeza del fémur,16 condición que sólo se observó en un paciente adulto. Quizá los niveles de hemoglobina mostrados por los niños SS en este estudio (rango 7.5 –9.7 g/dL) no sean lo suficientemente elevados como para producir un efecto deletéreo, o los niveles elevados de HbF en 38% de los niños, influyeron beneficiosamente. Por otro lado, aunque en los pacientes con hemoglobinopatía SS, normalmente la anemia es del tipo normocítica normocrómica, llama la atención el aumento del índice RDW, lo cual sugiere una deficiencia de hierro agregada; los pacientes evaluados pertenecen a los estratos más pobres de la población (clase obrera y marginal), y es una realidad en Venezuela y específicamente en el estado Sucre la alta incidencia de anemia ferropénica.17–19
Al respecto, se ha observado la eosinofilia como un patrón común en la población sucrense, y ha sido relacionada con parasitosis y cuadros asmáticos.20–22 En el estudio hematológico de los pacientes SS se observó eosinofilia, y en 71% de los expedientes clínicos se encontró registro de infección de origen parasitario principalmente del tipo helmíntico, resultando ésta la primera causa de hospitalización.
La frecuencia de crisis dolorosas o crisis vasooclusiva (CVO), fue menor a la descrita por otros autores en Cuba,23,24 y semejante a la reportada en Sicilia y Kuwait.25,26 Nagel4 expone que las principales causas precipitantes de las CVO en la AD, son el frío, el estrés y el ejercicio físico, entre otras. El Estado Sucre carece de los cambios climáticos que puedan exacerbar las crisis drepanocíticas. Se supone que las causas precipitantes de las crisis drepanocíticas en estos pacientes, fueron los cuadros infecciosos, ya que bajo estas condiciones, el pH de la sangre se vuelve más ácido y esto contribuye al aumento de la falciformación.
En los adultos SS, los valores elevados de VCM basal se correlacionaron significativamente con un mayor número de ingresos y de crisis drepanocíticas. Esta observación no fue detectada en trabajos similares,27 sin embargo, concuerda con la tesis de que los niveles altos de VCM aumentan la viscosidad sanguínea y retardan el flujo sanguíneo, induciendo la falciformación y bloqueo de la microvasculatura (crisis vasooclusiva); esto provoca isquemia en los tejidos y, en casos extremos, puede causar daño a los órganos vitales, lo cual constituye la principal causa de morbilidad y mortalidad de esta enfermedad.
Tanto niños como adultos SS, presentaron hepatomegalia y esplenomegalia, con características de establecimiento crónico desde los dos hasta los 36 años inclusive, lo que sugiere que la autoesplenectomía no es común en estos pacientes. Los resultados coinciden con los reportados en otras áreas geográficas.15
Por otro lado, en pacientes clínicos con (3–talasemia, niveles normales de A2 podrían estar presentes, tal como lo observaron Bravo et al.,28 lo que corrobora que el paciente reportado como AS con anemia microcítica hipocrómica y manifestaciones clínicas similares a los homocigotos, podría presentar también el fenotipo de S–β talasemia del tipo heterocigoto compuesto; al igual que el paciente AAF, con anemia microcítica hipocrómica, manifestaciones clínicas severas, y niveles de Hb A2 cercanos al 3%.
No se detectaron casos de la delección 3,7 de α–talasemia en esta investigación, sin embargo, no se descarta la presencia de esta mutación o de otros tipos de α–talasemia en la población, ya que Arends et al.,2 encontraron una alta frecuencia de –α3,7 –talasemia en pacientes portadores de al menos un gen βS provenientes de diferentes partes de Venezuela. Sin embargo, los resultados obtenidos en este estudio concuerdan con los reportados en Grecia y Sicilia, poblaciones donde la presencia de a–talasemia es rara y no coinciden con la anemia drepanocítica.25
Las manifestaciones clínicas y hematológicas de los pacientes con anemia drepanocítica y anemia microcítica hipocrómica aquí estudiados, pudiesen estar moduladas al menos por dos factores genéticos, como la persistencia de hemoglobina fetal y la presencia de β talasemia; aunque no se descartan otras variables genéticas, como, en el caso de los pacientes SS, los diferentes haplotipos del gen βS que presenten.29,30 Se descarta la –α3,7 –talasemia como factor genético modulador de estas manifestaciones.
Debido a que la principal causa de hospitalizaciones fue la infección gastrointestinal, se sugiere que la presencia de parasitosis intestinales podría ser un factor ambiental desencadenante de las crisis, por lo que se podrían evitar cuadros agudos de descompensación en estos pacientes realizando regularmente un control coproparasitológico.
AGRADECIMIENTOS
Al personal del Banco de Sangre del Hospital Universitario "Antonio Patricio de Alcalá", en especial a la Dra. Chelita Hernández, y al Laboratorio de Hemoglobinas Anormales "Dr. Tulio Arends", por su apoyo técnico durante la realización de este trabajo.
REFERENCIAS
1. Colombo B, Guerchicoff E, Martínez–Antuña G. Bases moleculares de las talasemias. Cuba: Editorial Pueblo y Educación; 1993, p. 267. [ Links ]
2. Arends A, Alvarez M, Velásquez D, Bravo M, Salazar R, Guevara J, Castillo O. Determination of b–globin gene cluster haplotypes and prevalence of a–thalassemia in sickle cell patients in Venezuela. Am J Haematol 2000; 64: 87–90. [ Links ]
3. El–Hazmi M, Al–Swailem A, Wasrsy A. Case studies on haemoglobin S heterozigotes with severe clinical manifestations. J Trop Pediatr 1990; 36: 223–9. [ Links ]
4. Nagel RL, Steinberg MH. Role of epistatic (modifier) genes in the modulation of the phenotypic diversity of sickle cell anemia. Pediatr Pathol Mol Med 2001; 20(2): 123–36 [ Links ]
5. Serjeant G. The geography of sickle cell disease: opportunities for understanding its diversity. Ann Saudi Med 1994; 14(3): 1–10. [ Links ]
6. Salazar R, Bejarano Y, González M, Arends A. Estratificación socioeconómica, parámetros hematológicos y variantes hemoglobínicas en escolares de tres poblaciones del Estado Sucre. Saber 2002; 14(1): 55–9. [ Links ]
7. Lehmann H, Hunstman R. Amsterdam. North–Holland. Men's haemoglobin; 1974, p. 567. [ Links ]
8. Robinson AR, Robson M, Harinson A, Zuelza W. A new technique for differentiation of haemoglobin. J Lab Clin Med 1957; 50: 745. [ Links ]
9. Betke K, Martin H, Schilicht I. Estimation small percentages of foetal haemoglobin. Nature 1959; 184: 1877–8. [ Links ]
10. Bernini L. Rapid determination of hemoglobin A2 by DEAE–cellulose chromatography. Biochem Genet 1969; 2: 305–8. [ Links ]
11. Sambrook J, Fritsch E, Maniatis T. Molecular cloning. 2nd Ed. NY, USA: Cold Spring Harbor Laboratory; 1989. [ Links ]
12. Jeampierre M. A rapid method for the purification of DNA from blood. Nucl Acid R 1987; 15(22): 9611. [ Links ]
13. Baysal E, Huisman T. Detection of common deletional a–thalassemia–2 determinants by PCR. Am J Hematol 1994; 46: 208–13. [ Links ]
14. Svarch E, Nordet I, Machín S, Fernández L, Mufiiz A, Wade M. La drepanocitosis en los cinco primeros años de vida. Sangre 1996; 41(1): 43–6. [ Links ]
15. Serjeant G, Serjeant B, Stephens R, Higgs D, Beckford M, Cook J, Thomas P. Determinants of haemoglobin level in steady–state homozygous sickle cell disease. Br J Haematol 1996; 92: 143–9. [ Links ]
16. Marouf R, Gupta R, Haider MZ, Al–Wazzan H, Adekile AD. Avascular necrosis of the femoral head in adult Kuwaiti sickle cell disease patients. Acta Haematol 2003; 110(1): 11–5. [ Links ]
17. Méndez–Castellano H, Bosh M,López BM. Proyecto Venezuela, Fundación para el desarrollo y crecimiento de la población venezolana, Fundacredesa. 1993. [ Links ]
18. Landaeta de Jiménez M, Fossi M, Cipriano M, del Busto K, García K, Escalona J, Méndez–Hernández H. El hambre y la salud integral. An Venez Nutr 2003; 16(2): 150–6. [ Links ]
19. Macías–Tomey C, Jiménez–Landaeta M, García MN, Hevia D, Layrisse M, Méndez–Castellano H. Crecimiento físico y estado nutricional antropométrico de hierro y vitamina A en escolares de Venezuela.
20. Smith LL, Thier S. Fisiopatología: principios biológicos de la enfermedad. 2nd Ed. Argentina: Edit. Med Panam; 1988, p. 1236. [ Links ]
21. Ramos L, Salazar R. Infestación parasitaria en niños de Cariaco, Estado Sucre, Venezuela y su relación con las condiciones socioeconómicas. Kasmera 1997; 25(3): 175–89. [ Links ]
22. Guilarte Del V, Jiménez D. Factores socioeconómicos y culturales que influyen sobre las parasitosis intestinales en escolares de Chacopata y Campoma. Saber 2002; 2(1): 138–43. [ Links ]
23. García T, Nordet I, Machín S, González A, Muñiz A, Martínez G, Wade M, Svarch E. Aportes al estudio de la drepanocitosis. Análisis clínico y hematológico en los primeros 5 años de la vida. Rev Cub Hematol Inmunol Hemoter 1999; 15(2): 96–104. [ Links ]
24. Espinosa E, Svarch E, Martínez G, Hernández P. La anemia drepanocítica en Cuba. Experiencia de 30 años. Rev Cub Hematol Inmunol Hemoter 1997; 12(2). [ Links ]
25. Schiliro G, Samperi P, Consalvo C, Gangarossa S, Testa R, Miraglia V, Lo Nigro L. Clinical, hematological, and molecular features in Sicilians with sickle cell disease. Hemoglobin 1992; 16(6): 469–80. [ Links ]
26. Adekile A, Hayder M. Morbidity, bs haplotype and a–globin gene patterns among sickle cell anemia patients in Kuwait. Acta Haematol 1996; 96: 150–4. [ Links ]
27. Olantunji P, Davies S. The predictive values of white cell count in assessing clinical severity of sickle cell anaemia in Afro–Caribbeans patients. Afr J Med Sci 2000; 29(1): 27–30. [ Links ]
28. Bravo M, Salazar R, Arends A, Alvarez M, Velazquez D, Guevara J, Castillo O. Detección de btalasemia mediante la técnica de Amplificación Refractaria de Sistemas de Mutaciones (ARMS–PCR). Inv Clin 1999; 40(3): 203–13. [ Links ]
29. Chang YC, Smith KD, Moore RD, Searjent GR, Dover GJ. An analysis of fetal hemoglobin variatin in sickle cell desease: The relative contributions of X–linked factor, â globin haplotype, á globin gen number, gender and age. Blood 1995; 85: 1111–7. [ Links ]
30. Salazar–Lugo R. La hemoglobina S en la población venezolana. Inv Clin 2004; 45(2): 109–92. [ Links ]

