










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de investigación clínica
On-line version ISSN 2564-8896Print version ISSN 0034-8376
Rev. invest. clín. vol.57 n.4 Ciudad de México Jul./Aug. 2005
Rincón del residente
Patogénesis de la hipertensión portal
Pathogenesis of portal hypertension
Aldo Montaño–Loza,* Judith Meza–Junco**
* Residente de tercer año de Gastroenterología.
** Residente de tercer año de Oncología. Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán.
Reimpresos:
Dr. Aldo Montaño–Loza
Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán.
Departamento de Gastroenterología.
Vasco de Quiroga No. 15, Col. Sección XVI, Tlalpan
14080, México, D.F.
Email: aldomontano@hotmail.com
Recibido el 5 de agosto de 2004.
Aceptado el 17 de febrero de 2005.
ABSTRACT
It is now well established that portal hypertension is not a purely mechanical phenomenon. Primary hemodynamic alterations develop in the hepatic and systemic circulatory systems; these alterations in combination with mechanical factors contribute to the development of portal hypertension. In the hepatic circulation, these hemodynamic alterations are characterized by vasoconstriction and impaired hepatic vasodilatory responses, whereas in the systemic circulation, particularly in the splanchnic bed, vessels are hyperemic with increased flow. Thus, an increase in intrahepatic resistance in conjunction with increased portal venous inflow, mediated through splanchnic dilation, contributes to the development of portal hypertension. The ensuing development of elevated flow and transmural pressure through collateral vessels from the hypertensive portal vasculature into the lower pressure systemic venous circulation accounts for many of the complications, such as bleeding esophageal varices, observed with portal hypertension. The importance of the primary vascular origin of portal hypertension is emphasized by the utility of current therapies aimed at reversing these hemodynamic alterations, such as nitrates, which reduce portal pressure through direct intrahepatic vasodilatation, and fi blockers and octreotide, which reduce splanchnic vasodilatation and portal venous inflow. New evidence concerning relevant molecular mechanisms of contractile signaling pathways in hepatic stellate cells and the complex regulatory pathways of vasoactive molecules in liver endothelial cells makes a better understanding of these processes essential for developing further experimental therapies for portal hypertension. This article examines the current concepts relating to cellular mechanism that underlie the hemodynamic alterations that characterize and account for the development of portal hypertension.
Key words. Portal hypertension. Hyperdynamic circulation. Hepatic and systemic circulatory systems. Hepatic venous pressure gradient.
RESUMEN
Actualmente está bien establecido que la hipertensión portal no es un fenómeno puramente mecánico. En esta entidad se presentan alteraciones hemodinámicas primarias en los sistemas circulatorios hepático y sistémico; estas alteraciones en combinación con factores mecánicos, contribuyen al desarrollo de la hipertensión portal. En la circulación hepática, las alteraciones hemodinámicas se caracterizan por vasoconstricción y una respuesta anómala a la vasodilatación, mientras que en la circulación sistémica, especialmente en el lecho esplácnico, los vasos están congestivos y con un flujo aumentado. Por lo tanto un incremento en las resistencias intrahepáticas asociado a un aumento del flujo venoso portal, mediado a través de la dilatación esplácnica, contribuyen al desarrollo de la hipertensión portal. La consecuencia del flujo y la presión transmural elevada a través de los vasos colaterales a partir de una vasculatura portal hipertensa hacia la circulación venosa sistémica con menor presión, conlleva a muchas de las complicaciones observadas en la hipertensión portal, como la hemorragia por várices esofágicas. La importancia del origen vascular primario de la hipertensión portal se basa en la utilidad de terapias actuales orientadas a revertir estas alteraciones hemodinámicas, como los nitratos que reducen la presión portal, a través de vasodilatación intrahepática directa y los P bloqueadores y octreótida, que reducen la vasodilatación esplácnica y el flujo venoso portal. Además, existen nuevas evidencias en relación con los mecanismos moleculares de vías de señalización contráctil de las células estelares hepáticas y complejas vías de regulación de sustancias vasoactivas en las células endoteliales hepáticas que han ayudado a entender mejor estos procesos esenciales para el desarrollo de terapias experimentales para la hipertensión portal. Este artículo revisa los conceptos actuales relacionados con los mecanismos celulares causales de las alteraciones hemodinámicas que caracterizan y condicionan el desarrollo de la hipertensión portal.
Palabras clave. Hipertensión portal. Circulación hiperdinámica. Sistema circulatorio hepático y sistémico. Gradiente de presión venosa hepático.
INTRODUCCIÓN
Etiología y fisiopatología de la hipertensión portal
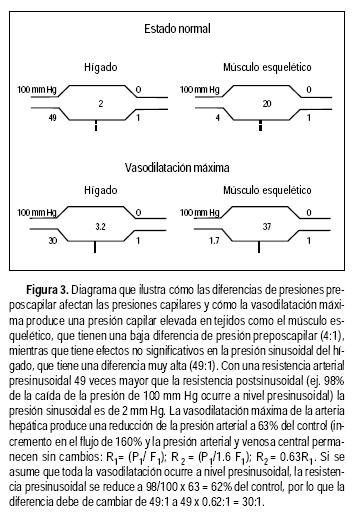
• Flujo sanguíneo en el hígado normal. El flujo sanguíneo hepático normal es de aproximadamente 1,500 mL/minuto, que representa de 15 a 20% del gasto cardiaco. Un tercio de este flujo lo provee la arteria hepática y dos tercios provienen del sistema venoso portal.1,2 A nivel de los sinusoides hepáticos el flujo sanguíneo arterial que tiene una presión elevada con altas concentraciones de oxígeno, se mezcla con el flujo sanguíneo venoso portal, que tiene una presión baja y una concentración pobre de oxígeno, pero es rico en nutrientes. La microcirculación hepática es peculiar debido a que la relación de la resistencia pre a postsinusoidal es de 49:1, a diferencia de la observada en el músculo esquelético, donde la relación de la resistencia pre a poscapilar es de 4:1.3
Después de perfundir los sinusoides, el flujo sanguíneo pasa de manera secuencial hacia las vénulas hepáticas, las venas hepáticas y la vena cava inferior, mientras una fracción de plasma entra al espacio de Disse y es drenado por los vasos linfáticos.
Una característica única de la microcirculación sinusoidal hepática es su baja presión de perfusión. Esta baja presión parece deberse a una elevada resistencia entre la región precapilar y poscapilar del hígado.4,5 Esto se debe a que los sinusoides están normalmente protegidos de la presión de perfusión portal y de sus fluctuaciones por un sitio presinusoidal de alta resistencia, probablemente localizado en las terminaciones venosas portales.6
Debido a que los sinusoides están cubiertos por un endotelio que carece de una membrana basal continua y que presentan múltiples fenestraciones de 50 a 200 nm de diámetro, el mantenimiento de una baja presión es crucial para sostener el equilibrio de la trasudación del fluido sinusoidal hacia el espacio de Disse.
Otra característica única de la circulación hepática, es la interrelación entre el flujo sanguíneo venoso portal y el de la arteria hepática. Cuando el flujo sanguíneo portal aumenta, el flujo sanguíneo de la arteria hepática disminuye; y cuando el flujo portal disminuye se incrementa el flujo arterial hepático. Este fenómeno se denomina "respuesta arterial de amortiguación hepática" y consiste en un reflejo vascular mediado por adenosina que asegura el mantenimiento de un estado relativamente constante de perfusión sinusoidal, en estados de cambios en el flujo portal, como ocurre normalmente con la ingesta de alimentos.1,2,7
El flujo sanguíneo del sistema vascular portal es dirigido por una diferencia de presiones o gradiente que se presenta a lo largo de todo el sistema. El gradiente de presión portal (Δ P), es la diferencia de presión entre el sistema venoso portal y el sistémico; y puede ejemplificarse como el resultado del producto del flujo venoso portal (Q) y las resistencias vasculares a este flujo (R), que se expresan por la ley de Ohm: Δ P = Q x R (Figura 1).
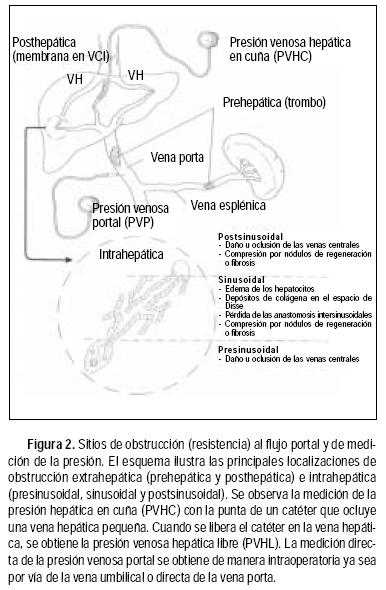
Normalmente, la presión portal es de 5 a 10 mm Hg (7–14 cm de agua) y puede ser modificada por la presión intraabdominal.8 La presión venosa hepática también puede verse afectada por la presión intraabdominal y representa la presión de llenado central. Para eliminar la posible contribución de la presión intraabdominal y de la presión venosa central sobre la presión portal, ésta se expresa como la diferencia de presión intrínseca entre los compartimentos venoso portal y el sistémico, esto es como el gradiente de presión portal (Δ P). Esta técnica consiste en medir la presión por medio de un catéter en cuña a nivel de la vena hepática con un abordaje yugular o femoral. Esta medición se denomina presión venosa hepática en cuña o enclavada (PVHC). Después de liberar la punta del catéter dentro de la vena hepática, se obtiene la presión venosa hepática libre (PVHL). El gradiente de presión venoso hepático (GPVH) se obtiene al restar el valor de la PVHL de la PVHC.9,10 La hipertensión portal (HTP) se define como un GPVH mayor de 6 mm Hg o también como una presión intraesplénica mayor de 15 mm Hg o una presión directa de la vena porta mayor de 21 mm Hg (30 cm de agua), cuando se determina durante cirugía3 (Figura 2).
Importancia de las resistencias vasculares
La cirrosis hepática por alcohol y por hepatitis viral comprende las principales causas de HTP en el mundo occidental.11,12 Existen otras causas de HTP que no se deben a cirrosis hepática y que son particularmente frecuentes en algunos países en desarrollo, como la esquistosomiasis.13 El esclarecimiento de los mecanismos fisiopatológicos de estas distintas entidades ha sido útil para la identificación de los diferentes niveles anatómicos de resistencia al flujo portal.
Las causas de HTP se clasifican convencionalmente de acuerdo con la localización del sitio de máxima resistencia al flujo portal14 (Cuadro 1). Las tres categorías principales son prehepática, intrahepática y posthepática. Así, la trombosis de la vena porta ejemplifica la HTP prehepática, mientras que una obstrucción a nivel de la vena cava inferior es típica de HTP posthepática. En el caso de la HTP intrahepática, ésta se subdivide en presinusoidal, sinusoidal y postsinusoidal. Es difícil caracterizar la HTP de acuerdo con el sitio intrahepático de mayor resistencia debido a la falta de equipo para medir directamente la presión de los sinusoides hepáticos. La mayor parte del conocimiento en esta área proviene de mediciones directas de la presión del sistema portal y de estimaciones indirectas de la presión intrasinusoidal a partir de la PVHC en correlación con las características anatomopatológicas de los hígados analizados.

Por ejemplo, tanto en la HTP prehepática y la intrahepática presinusoidal, la medición directa de la presión de la vena porta (PVP) está siempre elevada, en presencia de una PVHC y un GPVH normal. En la HTP intrahepática sinusoidal y postsinusoidal, la PVHC tiende a ser similar o igual que la medición directa de la PVP y el GPVH está aumentado de manera proporcional. En la HTP postsinusoidal, la PVHC está aumentada igual que la PVP y debido a que la PVHL está también aumentada, el GPVH usualmente es normal.58
La localización intrahepática de mayor resistencia suele establecerse más fácil en las causas de HTP no cirrótica.10,15 La enfermedad venooclusiva por el uso de alcaloides de pirrolizidina o quimioterapia, se caracteriza por una HTP intrahepática postsinusoidal evidente, mientras que la inflamación granulomatosa y fibrosis de la porta en la esquistosomiasis en etapas tempranas, es un ejemplo típico de HTP intrahepática presinusoidal. Sin embargo, algunos casos de HTP presinusoidal como la esquistosomiasis11,12 y la HTP idiopática,16,17 son más complicados con el curso del tiempo cuando la enfermedad progresa y genera patrones de resistencia mixtos.
El principal sitio de la resistencia vascular en la cirrosis ha sido difícil de establecer. La mayoría de las mediciones en cirrosis no alcohólica muestran una PVP mayor que la PVHC. Debido a que la PVHC es una estimación de la presión intrasinusoidal, este hallazgo se interpreta como un indicador de resistencia presinusoidal probablemente relacionado con la actividad inflamatoria o los cambios fibróticos en los tractos portales o por la presencia de anastomosis intersinusoidales que descomprimen parcialmente los sinusoides durante la medición de la presión en cuña.6
En los pacientes con cirrosis alcohólica la PVHC es igual a la PVP, lo que sugiere que el sitio de aumento en la resistencia incluye el sinusoide completo y que existe una menor descompresión por anastomosis intersinusoidales en esta enfermedad, posiblemente debido a una mayor fibrosis intrasinusoidal.9
Las alteraciones de la arquitectura hepática secundarias al desarrollo de septos fibróticos y nodulos de regeneración, conducen a cambios patológicos dentro de los sinusoides. Estos incluyen crecimiento del hepatocito, resultado de la acumulación de grasa y proteínas inducida por el alcohol, con compresión de los sinusoides hepáticos y obstrucción del flujo por depósitos de colágena en el espacio de Disse.9 Para localizar el sitio de mayor resistencia en la cirrosis alcohólica, hay que considerar que esta entidad no es homogénea. El sitio de mayor resistencia puede variar de acuerdo con el estadio, la actividad y las características morfológicas y patológicas predominantes. Por lo tanto, el hepatocito con edema puede ser un factor importante para el desarrollo de HTP sinusoidal parcialmente reversible en la hepatitis alcohólica aguda18 y la fibrosis perivenular con esclerosis hialina central son factores importantes en la resistencia postsinusoidal.6 Además, la formación de trombos en las venas hepáticas y portales de mediano y gran calibre19 pueden contribuir a los elementos de resistencia mixta pre y postsinusoidal.
El daño morfológico que ocurre en la enfermedad hepática crónica constituye, sin duda, el factor más importante para el incremento en la resistencia intrahepática. Sin embargo, existen estudios que muestran un papel dinámico de factores contráctiles que pueden aumentar el tono vascular. Los miofibroblastos son células contráctiles que se encuentran en cicatrices y áreas presinusoidales de hígados con cirrosis, pero que no se encuentran en los hígados sanos.20 Estas células parecen desarrollarse de células estelares activadas y presentan una respuesta contráctil a factores vasoconstrictores como las endotelinas,21 cuyos niveles están aumentados en sangre y tejido hepático en pacientes con cirrosis.17,22–24 La presión portal en los pacientes con cirrosis puede cambiar dependiendo en elementos contráctiles intrahepáticos y de la acción de factores vasoactivos en sangre. Si bien, las células estelares tienen un papel clave en la patogénesis de la fibrosis en el hígado con cirrosis, su contribución en la fluctuación de las resistencias intrahepáticas permanece sin esclarecerse.5,18
Para entender cómo la vasodilatación o la vasoconstricción pueden condicionar cambios importantes en el flujo sanguíneo en la mayoría de los lechos vasculares, pero no en el hígado, ejemplificaremos el efecto diferente de la vasodilatación en el músculo esquelético y en el hígado (Figura 3). En el músculo 80% de la resistencia del flujo deriva de los sitios precapilares. La diferencia de resistencia preposcapilar es de 4:1. La vasodilatación durante el ejercicio reduce esta diferencia aproximadamente 1.7:1, lo que resulta en una elevación de la presión capilar de cerca de 17 mm Hg y filtración importante de fluidos. La vasodilatación máxima de la arteria hepática producida por una infusión de isoproterenol produce un aumento en el flujo arterial de 160% de los valores controles.25 Se asume que todo el cambio de las resistencias vasculares ocurre a nivel precapilar, la diferencia de resistencia pre/postsinusoidal se reduciría a lo mucho a 30:1. Esta dilatación produciría un incremento en la presión sinusoidal menor de 1.5 mm Hg. El hecho de que existan dos suministros vasculares a nivel del sinusoide reduce los cambios hemodinámicos a nivel de uno de los vasos.
Con el desarrollo de venas colaterales durante la evolución de la HTP, la resistencia global del flujo venoso portal (Rp) es determinado por las resistencias paralelas de la resistencia vascular portohepática (RH) y la resistencia portocolateral (RC), de acuerdo con la siguiente relación: 1/Rp = 1/RH + 1/RC– Los vasos colaterales portosistémicos tienen gran cantidad de tejido muscular y por lo tanto presentan cambios en su diámetro en respuesta a sustancias vaso–activas, existiendo una mayor proporción de Rc en comparación con las RH sujeta a modificación.
Flujo sanguíneo portal
La contribución del aumento en el flujo venoso portal en la patogénesis de la HTP está sustentada por varios estudios clínicos y experimentales. Sin embargo, es importante considerar que los factores humorales, más allá de los efectos mecánicos son necesarios para la formación de los vasos colaterales 26,27
Entidades primarias con flujo portal elevado
Son poco frecuentes las entidades que condicionan un flujo elevado del sistema portal (fístulas arterio–portales, esplenomegalia secundaria a mielofibrosis o metaplasia mieloide). El desarrollo de HTP en pacientes con estas entidades refleja de manera invariable el efecto combinado del aumento del flujo portal y el incremento de las resistencias hepáticas.2,11 La HTP idiopática es una entidad comán en Asia y se pensaba que era un trastorno primario del bazo (de aquí su nombre antiguo, esplenomegalia tropical), con un aumento importante del flujo esplénico y por lo tanto del flujo esplenoportal. En realidad es una enfermedad de las tributarias preterminales de la vena porta (esclerosis hepatoportal),13 por lo que la HTP no se corrige con la esplenectomía.28 La importancia de la contribución del flujo portal elevado es utilizada para ilustrar el impacto clínico sobre la HTP, incluyendo la ascitis y las várices esofágicas, que pueden resolverse después de la normalización del flujo portal posterior al cierre de una fístula arterioportal.29
Algunas neoplasias pancreáticas pueden involucrar la vena porta o sus tributarias, lo que resulta en HTP extrahepática. Pueden desarrollarse vasos colaterales, usualmente con una dirección hepatopetal. Estos incluyen las venas gastroepiploicas, venas gástricas cortas, vena gástrica izquierda, venas cólicas izquierdas y comunicaciones esplenorrenales espontáneas.30
Paradoja de los vasos colaterales portosistémicos
Cuando la presión portal alcanza un valor crítico, se desarrollan vasos colaterales portosistémicos. En la cirrosis alcohólica es necesario un GPVH de 10 a 12 mm Hg para el desarrollo de várices esofágicas.31 Las venas colaterales son el resultado de la dilatación de conductos embrionarios o de la redirección del flujo dentro de venas existentes, más que la formación de nuevos vasos. De acuerdo a como se forman los vasos colaterales se espera que se descomprima el sistema portal y disminuya su presión. Paradójicamente, la extensión de las venas colaterales se correlaciona con el grado de presión portal.2 Esto obedece a un aumento compensatorio en el flujo portal o de las resistencias dentro del lecho colateral.23 La HTP se mantiene durante la formación de vasos colaterales por un incremento en el flujo portal y como consecuencia de la presión elevada, aunque exista fuga del flujo portal hacia estas colaterales.
En la HTP el flujo sanguíneo arterial esplácnico hacia el sistema venoso portal iguala el flujo portal hacia el hígado más el flujo portocolateral y en aproximadamente 10% de los pacientes el flujo de la vena porta puede estar revertido (flujo portal hepatofuga o retrógrado).32,33 Esta situación se presenta debido a que el flujo sanguíneo arterial hepático encuentra una gran resistencia para seguir su curso habitual o anterógrado a través de los sinusoides, en comparación con la resistencia ofrecida por las tributarias del sistema portal. Esta pérdida del flujo sanguíneo hepático o secuestro arterial hepático hacia las colaterales se asocia con un alto riesgo de deterioro de la función hepática y desarrollo de encefalopatía hepática.24,25,34,35
En este tópico es conveniente mencionar los cambios hemodinámicos relacionados con la sepsis, ya que es un modelo que unifica la información mencionada previamente. En términos prácticos, la designación de "hígado de choque" comprende dos etiologías importantes de daño hepático:
1. El choque hemorrágico o falla de la macrocirculación debido a otras causas.
2. El choque séptico. El hígado de choque también puede resultar de la falla orgánica múltiple y del síndrome de dificultad respiratoria aguda.
Es importante enfatizar que el hígado es el ánico órgano en donde dos sistemas venosos están conectados en serie. La circulación mesentérica–esplácnica es seguida por el sistema venoso portal y por lo tanto combina los efectos de la vasoconstricción esplácnica, la translocación bacteriana en el intestino y el flujo arterial mesentérico con la perfusión portal, que a su vez influye sobre el retorno venoso total que llega al corazón. En los pacientes sépticos con circulación hiperdinámica, el flujo hepatoesplénico también aumenta; sin embargo, el análisis de pacientes con y sin sepsis muestra resultados similares, por lo que la perfusión hepatoesplénica permanece estable independientemente de la etiología con una fracción aproximada de 25% del gasto cardiaco. El flujo sanguíneo durante el estado de choque también está regulado por la respuesta "amortiguadora" de la arteria hepática, que es capaz de asegurar el suministro de sangre por la dilatación de la arteria hepática en situaciones donde las presiones sistémicas medias sean menores de 50 mm Hg. En modelos animales esta regulación se observa principalmente en el choque hemorrágico, pero está alterada en el choque séptico. Cuando se administra endotoxina, la disminución del flujo portal debido a un incremento en las resistencias intrahepáticas no conlleva a un aumento en el flujo de la arteria hepática. Si bien, el flujo sanguíneo hepático global aumenta debido a un incremento en el gasto cardiaco, la regulación del flujo arterial y venoso está alterada en los estados de sepsis y este factor puede poner en riesgo de daño isquémico al hígado.36
Circulación hiperdinámica de la hipertensión portal
El aumento del flujo sanguíneo esplácnico en la HTP es el resultado de alteraciones hemodinámicas37,38 La asociación entre HTP y un estado hiperdinámico circulatorio fue descrita desde 1953.39 Sus características son un incremento del gasto cardiaco y una presión arterial disminuida. El gasto cardiaco elevado se debe a un aumento de la frecuencia cardiaca y del volumen circulante total. La disminución de la presión arterial se debe a una reducción en las resistencias vasculares secundaria a vasodilatación arterial periférica. La gravedad de las alteraciones circulatorias que acompañan a la cirrosis, se correlacionan con los índices clínicos de disfunción hepática.40 Si bien, estas mismas alteraciones pueden presentarse en pacientes con HTP no cirrótica,28 éstos suelen tener una circulación colateral portosistémica mayor, lo que sugiere que los cortocircuitos portosistémicos más que descompensar la función hepática constituyen el principal factor del estado circulatorio hiperdinámico.
Las consecuencias sistémicas de la circulación hiperdinámica en la enfermedad hepática terminal son complejas y representan una forma de disfunción orgánica múltiple.29 Se han descrito eventos adversos sobre la circulación renal, cerebral y pulmonar. En los pulmones la vasodilatación ocasiona hipoxemia arterial, que se observa en un tercio de los pacientes cirróticos en ausencia de enfermedad cardiorrespiratoria (síndrome hepatopulmonar). En este síndrome la disminución del tono vascular pulmonar condiciona una disociación sobre la difusión–perfusión, que es responsable de la hipoxemia.41,42
Los cambios hemodinámicos sistémicos observados en la HTP se explican por dos teorías opuestas, pero no mutuamente excluyentes. La teoría de la vasodilatación periférica,43 donde factores asociados con la cirrosis o los cortocircuitos portosistémicos causan vasodilatación arterial, principalmente a nivel esplácnico. Como consecuencia de la vasodilatación periférica aumenta el gasto cardiaco por la reducción de la poscarga, lo que produce un estado de circulación hiperdinámica. Una teoría alterna propone que un estímulo primario (reflejo hepatorrenal) para la retención de sodio y agua es una consecuencia directa de la HTP.28,29,44 Como resultado de la retención de agua y sodio aumenta el volumen sanguíneo y el gasto cardiaco. De acuerdo con esta teoría, la vasodilatación periférica ocurre como un fenómeno de adaptación para estos eventos tempranos.
Factores vasoactivos en la patogénesis de la hipertensión portal
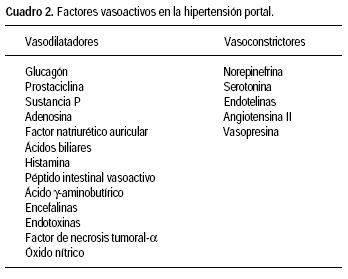
Varios factores vasoactivos humorales y autocrinos juegan un papel importante en la HTP. Mediadores vasoconstrictores y vasodilatadores han sido implicados y actúan sobre la circulación esplácnica y sistémica promoviendo un aumento de las resistencias vasculares intrahepáticas y de los lechos portocolaterales (Cuadro 2). En los pacientes con cirrosis hepática existe un aumento del tono nervioso simpático, ya que los niveles de epinefrina están aumentados. Sin embargo, parece existir una atenuación del efecto de los neurotransmisores simpáticos como resultado de una disminución de la densidad de receptores adrenérgicos y un antagonismo postreceptor por los factores vasodilatadores.45–47 Algunos estudios muestran que la perfusión de suero de animales con HTP a animales normales, condiciona vasodilatación arteriolar, por lo que se consideró que había vasodilatadores humorales que podían ser transferibles.3
Desde entonces, se han estudiado múltiples mediadores vasoactivos responsables de la vasodilatación esplácnica que se acompaña de aumento en el flujo de la vena porta.36,48 Al parecer los vasodilatadores endógenos normalmente se encuentran presentes en la sangre portal y son depurados por el hígado. Sin embargo, en la cirrosis éstos pueden evitar la depuración pasando por cortocircuitos portosistémicos o por alteración del metabolismo hepatocelular.
Varios péptidos gastrointestinales parecen tener efecto vasodilatador en la HTP. Los niveles séricos de glucagón están aumentados en modelos experimentales de HTP y en pacientes con cirrosis hepática.3,49,50 El glucagón afecta la respuesta vascular sistémica a la norepinefrina39 y también se ha observado una reducción del flujo sanguíneo esplácnico con la infusión antisuero específico de glucagón.51,52 Sin embargo, esta disminución del flujo esplácnico no se acompaña de disminución en la vasodilatación sistémica. Además, otros estudios no han encontrado correlación entre los niveles circulantes de glucagón y la vasodilatación arterial.53 La infusión de somatostatina o su análogo sintético octreótida, disminuye la liberación de glucagón y produce vasoconstricción en la circulación esplácnica y sistémica.54 Debido a que la somatostatina también inhibe la liberación de otros péptidos vasodilatadores, como la sustancia P, péptido intestinal vasoactivo (PIV) y péptido genéticamente relacionado con calcitonina (PRGC), es probable que sus efectos sobre la circulación en la HTP sean mediados de forma aditiva o de manera independiente del glucagón. Además, la somatostatina tiene un efecto vasoconstrictor directo sobre el músculo liso vascular.36 Aún no se comprende totalmente el papel del glucagón como mediador de vasodilatación sistémica, pero con la evidencia disponible parece ser responsable de aproximadamente 30 a 40% de la vasodilatación esplácnica en la HTP.35
Parece que el blanco molecular del glucagón para mediar los cambios de tono del músculo liso vascular, es el canal de K+ dependiente de ATP.55 Incluso en los pacientes con cirrosis que reciben glucosa, la administración de glibenclamida atenáa la vasodilatación esplácnica y renal inducida por glucosa. Además, la glibenclamida per se promueve una disminución del índice cardiaco; por lo que estos factores deben de ser considerados cuando se administra glibenclamida a un paciente con cirrosis y diabetes mellitus tipo 2.56
Los factores vasoactivos producidos por el endotelio vascular también tienen un papel en la HTP. El óxido nítrico (ON) es un potente vasodilatador endógeno producido en varios tejidos de manera constitutiva por la enzima vascular endotelial ON sintasa (eONS) y de manera inducible a partir del aminoácido L–arginina por la sintasa inducible de ON (iONS).37 En el parénquima hepático el ON es producido de manera constitutiva por la eONS y en las células no parenquimatosas por la iONS, después de su inducción por citocinas y endotoxinas. Parece que una producción excesiva de ON por la eONS está asociada a la disminución de las resistencias vasculares esplácnicas y periféricas en la HTP.37,57–61 La administración de antagonistas del ON en animales con HTP induce vasoconstricción sistémica y esplácnica mejorando el estado de circulación hiperdinámica43,44 Además, la inhibición de la síntesis de ON corrige parcialmente la falta de respuesta a los vasoconstrictores, que es característica de la HTP.37 Los pacientes con cirrosis hepática presentan concentraciones séricas y urinarias elevadas de nitritos y nitratos (productos finales de la oxidación de ON), lo que sugiere que el ON es un factor importante para el desarrollo de las alteraciones circulatorias;62,63 sin embargo, la inhibición del ON atenúa pero no revierte el estado de circulación hiperdinámica de la HTP.43,44 Además, un estudio en animales donde se inhibió la síntesis de ON de manera crónica, retardó pero no impidió la vasodilatación esplácnica.64,65 Estos estudios sugieren que hay otros factores además del ON que están involucrados en el fenómeno de vasodilatación asociada a la circulación hiperdinámica en la HTP.
Las prostaglandinas parecen tener un lugar en el desarrollo de la circulación hiperdinámica.66,67 Los niveles de prostaciclina están elevados en la sangre portal de ratas con HTP,48 mientras que los pacientes con cirrosis tienen un aumento en los niveles de prostaciclinas sistémicas y en sangre de la vena porta. Además, los niveles de prostaciclina en sangre portal se correlacionan con el grado de elevación en su presión.48,49
Estudios experimentales han mostrado que la prostaglandina E2 (PGE2) no parece tener un papel en los cambios hemodinámicos observados durante el desarrollo de la HTP. Sin embargo, la producción de PGE2 a nivel pulmonar parece estar incrementada como respuesta al aumento de la presión portal.68 Además, la inhibición de la biosíntesis de prostaglandinas con indometacina, mejora la circulación hiperdinámica y la presión portal en pacientes con cirrosis hepática de HTP.35,–69–71
Otros vasodilatadores han sido estudiados, como los ácidos biliares, histamina, adenosina y la sustancia P, sin encontrar evidencia convincente de que contribuyan al estado hiperdinámico de la HTP.
Resistencia vascular intrahepática
Parece que la contracción de la célula estelar contribuye con el incremento de las resistencias intrahepáticas en la HTP. Tanto los vasodilatadores como los vasoconstrictores pueden modular las resistencias vasculares intrahepáticas, a través de relajación y contracción de estas células o de otros elementos contráctiles, como los esfínteres vasculares hepáticos.72,73
Las endotelinas (ET) son una familia de al menos tres péptidos de 21–aminoácidos (ET–1, ET–2 y ET–3),74 que son agonistas contráctiles de las células estelares.50 La ET–1 es liberada por las células endoteliales y es estimulada por la epinefrina y la angiotensina II. A su vez, la ET–1 incrementa los niveles de dichos vasopresores que junto con la vasopresina aumentan la resistencia vascular intrahepática. La infusión de ET–1 aumenta la presión portal y promueve el cierre de las fenestraciones endoteliales,75 contribuyendo al fenómeno de "capilarización" que se presenta en la HTP. Además, la ET–1 parece favorecer el desarrollo de fibrosis hepática y su bloqueo farmacológico podría ser una opción terapéutica para las enfermedades hepáticas crónicas y sus complicaciones.76
Los niveles plasmáticos y hepáticos de ET están aumentados en los pacientes con cirrosis, principalmente aquellos con ascitis.19,20,77 El mecanismo responsable del incremento de ET no es del todo claro, pero parece deberse a una estimulación por el factor de crecimiento transformante–β el factor de necrosis tumoral–α (TNF–α) y por el estrés mecánico del flujo.21 El ON también ha sido implicado en la modulación de las resistencias intrahepáticas y parece tener un papel en la regulación de la circulación portal intrahepática. Parece existir un estado de deficiencia relativa de ON en el paciente con cirrosis, como resultado de disfunción endotelial sinusoidal. La deficiencia de ON en el paciente con cirrosis puede tener consecuencias importantes, resultado no sólo de la alteración en la relajación, sino también de la pérdida de efectos protectores del ON, incluyendo mecanismos antifibrogénicos y antitrombóticos.37
Resistencia porto–colateral
En la HTP avanzada, la circulación colateral lleva cerca de 90% de la sangre que entra en el sistema portal. Bajo estas circunstancias es obvio que las resistencias de los vasos colaterales tienen un impacto directo sobre la presión portal. Los factores que modulan la resistencia colateral no están bien estudiados. Estudios recientes sugieren que el ON es importante para la formación de colaterales portosistémicas y para la reducción de las resistencias de estos vasos. La serotonina tiene efecto vasoconstrictor mediado por los receptores de 5–hidroxitriptamina (5–HT2) en la circulación venosa esplácnica. La administración de bloqueadores selectivos de receptores 5–HT2 (ketanserina y ritanserina) causan una disminución significativa en la presión portal, sin modificar el flujo portal, lo que sugiere un efecto sobre las resistencias portocolaterales responsable de esta elevación en la presión portal.35,36
La flora intestinal y la circulación hiperdinámica
Estudios realizados en ratas con cirrosis y ascitis, han sido útiles para ejemplificar la secuencia de eventos a partir de la translocación bacteriana y endotoxinas, con la liberación de citocinas y la sobreproducción de ON y el subsiguiente desarrollo del estado de circulación hiperdinámica.78 En el primer estudio79 las ratas con cirrosis y ascitis se dividieron de acuerdo con la presencia o ausencia de translocación bacteriana. Todas las ratas con ascitis presentaron una pobre respuesta a los vasoconstrictores en el lecho esplácnico y una presión arterial media baja, en comparación con las ratas normales; sin embargo, estas alteraciones fueron significativamente mayores en las ratas que presentaban translocación bacteriana. Los cambios hemodinámicos se correlacionaron con un aumento en las concentraciones de ON y TNF–α, por lo que este estudio demostró que la translocación bacteriana empeora el estado de circulación hiperdinámica en la cirrosis hepática. Estos hallazgos fueron similares a los reportados en un estudio subsiguiente,80 que mostró que la translocación bacteriana se asocia con la presencia de endotoxinas sistémicas y en ganglios linfáticos mesentéricos, y se correlaciona con un incremento en los niveles séricos de ON. Si bien, el TNF–β y el lipopolisacárido (LPS) son estimuladores de la iONS, ambos estudios demostraron que el aumento del ON fue derivado de la eONS. Otra vía por la cual el TNF–P y el LPS pueden favorecer la sobreproducción de ON, parece ser a través de la estimulación de la GTP–ciclohidrolasa y la síntesis de la tetrahidrobiopterina, que activa la eONS, sin ejercer efecto alguno sobre la iONS. Además, existe evidencia que muestra que la descontaminación selectiva del intestino con norfloxacina parece atenuar el desarrollo del estado de circulación hiperdinámica en los pacientes con cirrosis.81 83 Sin embargo, es importante señalar que el papel de los productos bacterianos en el aumento del ON y el empeoramiento del estado de circulación hiperdinámica parece estar limitado a un subgrupo de pacientes con cirrosis descompensada y en estados avanzados de la enfermedad.
CONCLUSIONES
El entendimiento de la fisiopatología de la HTP ha evolucionado considerablemente en los últimos años a partir de conceptos simples de obstrucción del flujo venoso portal por cambios en la arquitectura del hígado y vasodilatación esplácnica y sistémica, a un estado hemodinámico que se ve regulado por componentes neurales, celulares y humorales que actúan de manera endocrina, paracrina y autocrina. Por lo tanto, debe enfatizarse que la HTP no es consecuencia de un fenómeno puramente mecánico. En esta entidad se presentan alteraciones hemodinámicas primarias de los sistemas circulatorios hepático y sistémico, que en combinación con factores mecánicos contribuyen a su desarrollo. Las terapias futuras deberán tener como blanco varias citocinas y sustancias vasoactivas y no enfocarse exclusivamente en sus efectos a nivel hepático, debido a los diversos efectos hemodinámicos de estos agentes sobre la circulación sistémica, portal y colateral.
REFERENCIAS
1. Huet PM, Pomier–Layrargues G, Villeneuve JP, et al. Intrahepatic circulation in liver disease. Semin Liver Dis 1986; 6: 277–86. [ Links ]
2. Boyer TD. Portal hypertensive hemorrhage: pathogenesis and risk factors. Semin Gastrointest Dis 1995; 6: 125–33. [ Links ]
3. Kapoor D, Sarin SK. Pathophysiology of portal hypertension. J Gatroenterol Hepatol 2002; 17: S482–S487. [ Links ]
4. Benoit JN, Granger DN. Splanchnic hemodynamics in chronic portal hypertension. Semin Liver Dis 1986; 6: 287–98. [ Links ]
5. Lebrec D, Moreau R. Pathogenesis of portal hypertension. Eur J Gastroenterol Hepatol 2001; 13: 309–11. [ Links ]
6. Ekataksin W, Kaneda K. Liver microvascular architecture: an insight into the pathophysiology of portal hypertension. Semin Liver Dis 1999: 19: 359–82. [ Links ]
7. Groszmann RJ, Atterbury CE. The pathophysiology of portal hypertension. Semin Liver Dis 1982; 2: 177–86. [ Links ]
8. Sandblom P. The history of portal hypertension. J R Soc Med 1993; 86: 544–6. [ Links ]
9. Polio J, Groszmann RJ. Hemodynamic factors involved in the development and rupture of esophageal varices: a pathophysiologic approach to treatment. Semin Liver Dis 1986; 6: 318–31. [ Links ]
10. Lebrec D, Bataille C, Bercoff E, et al. Hemodynamic changes in patients with portal venous obstruction. Hepatology 1983; 3: 550–3. [ Links ]
11. Garcia–Tsao G. Current management of the complications of cirrhosis and portal hypertension: varices and variceal hemorrhage, ascites and spontaneous bacterial peritonitis. Gastroenterology 2001; 120: 726–48. [ Links ]
12. Mendez–Sanchez N, Aguilar–Ramirez JR, Reyes A, et al. Etiology of liver cirrhosis in Mexico. Ann Hepatol 2004; 3: 30–3. [ Links ]
13. Raia S, Mies S, Macedo AL. Portal hypertension in schistosomiasis. Clin Gastroenterol 1985; 14: 57–82. [ Links ]
14. Lebrec D, Benhamou JP. Noncirrhotic intrahepatic portal hypertension. Semin Liver Dis 1986; 6: 332–40. [ Links ]
15. Da Silva LC, Carrilho FJ. Hepatosplenic schistosomiasis: Pathophysiology and treatment. Gastroenterol Clin North Am 1992; 21: 163–77. [ Links ]
16. Ludwig J, Hashimoto E, Obata H, et al. Idiopathic portal hypertension. Hepatology 1993; 17: 1157–62. [ Links ]
17. Hillaire A, Bonte E, Denninger MH, et al. Idiopathic non–cirrhotic intrahepatic portal hypertension in the West: a re–evaluation in 28 patients. Gut 2002; 51: 275–80. [ Links ]
18. Benhamou JP, Valla DC. Intrahepatic portal hypertension. In: Bircher J, Benhamou JP, Mclntyre N, et al, eds. Oxford textbook of clinical hepatology, 2nd edn. Oxford: Oxford University Press; 1999, p. 661–70. [ Links ]
19. Wanless IR, Wong F, Blendis LM, et al. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology 1995; 21: 1238–47. [ Links ]
20. Rockey DC. Cellular pathophysiology of portal hypertension and prospects for management with gene therapy. Clin Liver Dis 2001; 5: 851–65. [ Links ]
21. Pinzani M, Gentilini P. Biology of hepatic stellate cells and their possible relevance in the pathogenesis of portal hypertension in cirrhosis. Semin Liver Dis 1999; 19: 397–410. [ Links ]
22. Moller S, Bendtsen F, Heriksen JH. Vasoactive substances in the circulatory dysfunction of cirrhosis. Scand J Clin Lab Invest 2001; 61: 421–9. [ Links ]
23. Alam I, Bass NM, Bacchett P, et al. Hepatic tissue levels of endothelin–1 correlate with severity of chronic liver disease and ascites. Am J Gastroenterol 2000; 95: 199–203. [ Links ]
24. Reynaert H, Thompson MG, Thomas T, et al. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut 2002; 50: 571–81. [ Links ]
25. Lautt WW. Hepatic vasculature: a conceptual review. Gastroenterology 1977; 73: 1163–69. [ Links ]
26. Chan CC, Wang SS, Lee FY, et al. Effects of endothelin–1 on portal–systemic collaterals of common bile duct–ligated cirrhotic rats. Eur J Clin Invest 2004; 34: 290–6. [ Links ]
27. Fernandez M, Vizzutti F, Garcia–Pagan JC, et al. Anti–VEGF receptor–2 monoclonal antibody prevents portal–systemic collateral vessel formation in portal hypertensive mice. Gastroenterology 2004; 126: 886–94. [ Links ]
28. Matsubara S, Ouchi K, Matsuno S. Portal venous pressure following splenectomy in patients with portal hypertension of differing etiology. Eur Surg Res 1992; 24: 372–7. [ Links ]
29. Vauthey JN, Tomczak RJ, Helmberger T, et al. The arterioportal fistula syndrome: clinicopathologic features, diagnosis, and therapy. Gastroenterology 1997; 113: 1390–401. [ Links ]
30. Kamel IR, Lawler LP, Corl FM, et al. Patterns of collateral pathways in extrahepatic portal hypertension as demonstrated by multidetector row computed tomography and advanced image processing. J Comput Assist Tomogr 2004; 28: 469–77. [ Links ]
31. Roberts LR, Kamath PS. Pathophysiology of variceal bleeding. Gastrointest Endose Clin North Am 1999; 9: 167–74. [ Links ]
32. Rector WG Jr., Hoefs JC, Hossack KF, et al. Hepatofugal portal flow in cirrhosis: observations on hepatic hemodynamics and the nature of the arterioportal communications. Hepatology 1988; 8: 16–20. [ Links ]
33. Gaiani S, Bolondi L, Li Bassi S, et al. Prevalence of spontaneous hepatofugal portal flow in liver cirrhosis: clinical and endoscopic correlation in 228 patients. Gastroenterology 1991; 100: 160–7. [ Links ]
34. Sugita S, Ohnishi K, Saito M, et al. Splanchnic hemodynamics in portal hypertensive dogs with portal fibrosis. Am J Physiol 1987; 252: G748–G754. [ Links ]
35. Nishida O, Moriyasu F, Nakamura T, et al. Interrelationship between splenic and superior mesenteric venous circulation manifested by transient splenic arterial occlusion using a balloon catheter. Hepatology 1987; 7: 442–6. [ Links ]
36. Strassburg CP. Gastrointestinal disorders of the critically ill. Shock liver. Best Pract Res Clin Gastroenterol 2003; 17: 369–81. [ Links ]
37. Ready J, Rector WG Jr. Systemic hemodynamic changes in portal hypertension. Semin Gastrointest Dis 1995; 6: 134–9. [ Links ]
38. Groszmann RJ. Hyperdynamic circulation of liver disease 40 years later: pathophysiology and clinical consequences. Hepatology 1994; 20: 1359–63. [ Links ]
39. Abelmann WH. Hyperdynamic circulation in cirrhosis: a historical perspective. Hepatology 1994; 20: 1356–8. [ Links ]
40. Meng HC, Lin HC, Tsai YT, et al. Relationships between the severity of cirrhosis and haemodynamic values in patients with cirrhosis. J Gastroenterol Hepatol 1994; 9: 148–53. [ Links ]
41. Lange PA, Stoller JK. The hepatopulmonary syndrome. Ann Intern Med 1995; 122: 521–9. [ Links ]
42. Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004; 363: 1461–68. [ Links ]
43. Schrier RW, Arroyo V, Bernardi M, et al. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology 1988; 8: 1151–7. [ Links ]
44. Rector WG Jr., Robertson AD, Lewis FW, et al. Arterial underfilling does not cause sodium retention in cirrhosis. Am J Med 1993; 95: 286–95. [ Links ]
45. García–Pagan JC, Bosch J, Rodés J. The role of vasoactive mediators in portal hypertension. Semin Gastrointest Dis 1995; 6: 140–7. [ Links ]
46. García–Pagan JC, Escorsell A, Moitinho E, et al. Influence of pharmacological agents on portal hemodynamics: basis for its use in the treatment of portal hypertension. Semin Liver Dis 1999; 19: 427–38. [ Links ]
47. Colle IO, De Vriese AS, Van Vlierberghe HR, et al. Vascular hyporesponsiveness in the mesenteric artery of anaesthetized rats with cirrhosis and portal hypertension: an in–vivo study. Eur J Gastroenterol Hepatol 2004; 16: 139–45. [ Links ]
48. Wiest R, Groszmann RJ. Nitric oxide and portal hypertension: its role in the regulation of intrahepatic and splanchnic vascular resistance. Semin Liver Dis 1999; 19: 411–26. [ Links ]
49. Silva G, Navasa M, Bosch J, et al. Hemodynamic effects of glucagon in portal hypertension. Hepatology 1990; 11: 668–73. [ Links ]
50. Pizcueta MP, Casamitjana R, Bosch J, et al. Decreased systemic vascular sensitivity to norepinephrine in portal hypertensive rats: role of hyperglucagonism. Am J Physiol 1990; 258: G191–G195. [ Links ]
51. Benoit JN, Zimmermann B, Preman AJ, et al. Role of glucagon in splanchnic hyperemia of chronic portal hypertension. Am J Physiol 1986; 251: G674–G647. [ Links ]
52. Tsui CP, Sung JJ, Leung FW. Role of acute elevation of portal venous pressure by exogenous glucagon on gastric mucosal injury in rats with portal hypertension. Life Sci 2003; 73: 1115–29. [ Links ]
53. Sikuler E, Groszmann RJ. Hemodynamic studies in long–and short–term portal hypertensive rats: the relation to systemic glucagon levels. Hepatology 1986; 6: 414–8. [ Links ]
54. Albulos A, Colombato LA, Lee FY. Octreotide ameliorates vasodilation and Na+ retention in portal hypertensive rats. Gastroenterology 1993; 104: 575–9. [ Links ]
55. Cao K, Tang G, Hu D, Wang R. Molecular basis of ATP–sensitive K+ channels in rat vascular smooth muscles. Biochem Biophys Res Commun 2002; 296: 463–9. [ Links ]
56. Moreau R, Chagneau C, Heller J, et al. Hemodynamic, metabolic and hormonal responses to oral glibenclamide in patients with cirrhosis receiving glucose. Scand J Gastroenterol 2001; 36: 303–8. [ Links ]
57. Bomzon A, Blendis LM. The nitric oxide hypothesis and the hyperdynamic circulation in cirrhosis. Hepatology 1994; 20: 1343–50. [ Links ]
58. Sogni P, Moreau R, Gadano A, et al. The role of nitric oxide in the hyperdynamic circulatory syndrome associated with portal hypertension. J Hepatol 1995; 23: 218–24. [ Links ]
59. Lopez–Talavera JC, Merrill WW, Groszmann RJ. Tumor necrosis factor alpha: a major contributor to the hyperdynamic circulation in prehepatic portal–hypertensive rats. Gastroenterology 1995; 108: 761–7. [ Links ]
60. Theodorakis NG, Wang YN, Skill NJ, et al. The role of nitric oxide synthase isoforms in extrahepatic portal hypertension: studies in gene–knockout mice. Gastroenterology 2003; 124: 1500–8. [ Links ]
61. Wang JJ, Gao GW, Gao RZ, et al. Effects of tumor necrosis factor, endothelin and nitric oxide on hyperdynamic circulation of rats with acute and chronic portal hypertension. World J Gastroenterol 2004; 10: 689–93. [ Links ]
62. Guarner C, Soriano G, Tomas A, et al. Increased serum nitrite and nitrate levels in patients with cirrhosis: relationship to endotoxemia. Hepatology 1993; 18: 1139–43. [ Links ]
63. Tsai MH, Iwakiri Y, Cadelina G, et al. Mesenteric vasoconstriction triggers nitric oxide overproduction in the superior mesenteric artery of portal hypertensive rats. Gastroenterology 2003; 125: 1452–61. [ Links ]
64. García–Pagan JC, Fernández M, Bernadich C, et al. Effects of continued nitric oxide inhibition on the portal hypertensive syndrome after portal vein stenosis in the rat. Am J Physiol 1994; 30: 984–90. [ Links ]
65. Grange JD, Amiot X. Related articles, links nitric oxide and renal function in cirrhotic patients with ascites: from physiopathology to practice. Eur J Gastroenterol Hepatol 2004; 16: 567–70. [ Links ]
66. Guarner C, Soriano G. Prostaglandin and portal hypertension. Prostaglandins Leukot Essent Fatty Acids 1993; 48: 203–6. [ Links ]
67. Guarner C, Soriano G, Such J, et al. Systemic prostacyclin in cirrhotic patients. Relationship with portal hypertension and changes after intestinal decontamination. Gastroenterology 1992; 102: 303–9. [ Links ]
68. Ackerman Z, Karmeli F, Rachmilewitz D. Longitudinal prostaglandin E2 generation in various organs during evolution of experimental portal hypertension. Prostaglandins Leukot Essent Fatty Acids 2002; 67: 197–201. [ Links ]
69. Birney Y, Redmond EM, Sitzmann JV, et al. Eicosanoids in cirrhosis and portal hypertension. Prostaglandins Other Lipid Mediat 2003; 72: 3–18. [ Links ]
70. Yokoyama Y, Toth B, Kitchens WC, et al. Role of thromboxane in producing portal hypertension following trauma–hemorrhage. Am J Physiol Gastrointest Liver Physiol 2003; 285: G1293–G1299. [ Links ]
71. Gonzalez–Garcia M, Albillos–Martinez A. Pharmacological modulation of portal hypertension syndrome: current status and future prospects. Gastroenterol Hepatol 2004; 27(Suppl 1): 1–7. [ Links ]
72. Kawada N, Tran–Thi TA, Klein H, et al. The contraction of hepatic stellate cells stimulated with vasoactive substances. Possible involvement of endothelin–1 and nitric oxide in the regulation of sinusoidal tonus. Eur J Biochem 1993; 213: 815–23. [ Links ]
73. Zhang JX, Pegoli W, Clemens MG. Endothelin–1 induces direct constriction of hepatic sinusoids. Am J Physiol 1994; 266: G624–G632. [ Links ]
74. Levin ER. Endothelins. N Engl J Med 1995; 333: 356–63. [ Links ]
75. Reichen J, Gerbes AL, Steiner MJ, et al. The effect of endothelin and its antagonist Bosentan on hemodynamics and microvascular exchange in cirrhotic rat liver. J Hepatol 1998; 28: 1020–30. [ Links ]
76. Thirunavukkarasu C, Yang Y, Subbotin VM, et al. Endothelin receptor antagonist TAK–044 arrests and reverses the development of carbon tetrachloride induced cirrhosis in rats. Gut 2004; 53: 1010–19. [ Links ]
77. Ginés P, Cardenas A, Arroyo V, et al. Management of cirrhosis and ascites. N Engl J Med 2004; 350: 1646–54. [ Links ]
78. Garcia–Tsao G, Weist R. Gut microflora in the pathogenesis of the complications of cirrhosis. Best Pract Res Clin Gastroenterol 2004; 18: 353–72. [ Links ]
79. Wiest R, Das S, Cadelina G, et al. Bacterial translocation to lymph nodes of cirrhotic rats stimulates eNOS–derived NO production and impairs mesenteric vascular contractility. J Clin Invest 1999; 104: 1223–33. [ Links ]
80. Wiest R, Tsai MH, Garcia–Tsao G, et al. Bacterial translocation up–regulates GTP–cyclohydrolase I in mesenteric vasculature of cirrhotic rats. Hepatology 2003; 38: 1508–15. [ Links ]
81. Albulos A, de la Hera A, Gonzalez M, et al. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology 2003; 37: 208–17. [ Links ]
82. Chin–Dusting JP, Rasaratnam B, Jennings JL, et al. Effect of fluoroquinolone on the enhanced nitric oxide–induced peripheral vasodilation seen in cirrhosis. Ann Intern Med 1997; 127: 985–8. [ Links ]
83. Rabiller A, Nunes H, Lebrec D, et al. Prevention of gram–negative translocation reduces the severity of hepatopulmonary syndrome. Am J Resp Crit Care Med 2002; 166: 514–17. [ Links ]

