










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de investigación clínica
versión On-line ISSN 2564-8896versión impresa ISSN 0034-8376
Rev. invest. clín. vol.57 no.3 Ciudad de México may./jun. 2005
Rincón del residente
Daño pulmonar agudo asociado a ventilación mecánica
Ventilator associated acute lung injury
Silvio Antonio Ñamendys–Silva,* Juan Gabriel Posadas–Calleja**
* División de Medicina Crítica.
** Unidad de Terapia Intensiva. División de Medicina Crítica. Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán.
Reimpresos:
Dr. Silvio Antonio Ñamendys–Silva
Departamento de Medicina Crítica,
Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán.
Vasco de Quiroga 15, Tlalpan, Sección XVI,
14080, México, D.F.
Correo electrónico: tony75ni@msn.com
Recibido el 9 de marzo de 2004.
Aceptado el 11 de octubre de 2004.
ABSTRACT
Mechanical ventilation plays a central role In the critical care setting; but its use is closely related with some life threatening complications as nosocomial pneumonia and low cardiac performance. One of the most severe complications is called ventilator–associated lung injury (VALI) and it includes: Barotrauma, volutrauma, atelectrauma, biotrauma and oxygen–mediated toxic effects and it is related with an inflammatory response secondary to the stretching and recruitment process of alveoli within mechanical ventilation. The use of some protective ventilatory strategies has lowered the mortality rate 10% approximately.
Key words. Ventilator–associated lung injury. PEEP. Low tidal volume. Protective ventilatory strategies.
RESUMEN
La importancia de la asistencia mecánica ventilatoria (AMV) en la Unidad de Cuidados Intensivos (UCI) es indiscutible; sin embargo, su uso está ligado con complicaciones como neumonía nosocomial y deterioro del rendimiento cardiaco, que en algunas ocasiones ponen en peligro la vida del enfermo. Una de las complicaciones más graves es el daño pulmonar asociado a la ventilación mecánica (DPVM). El DPVM se caracteriza por la presencia de edema pulmonar rico en proteínas. Se recomienda establecer cierto número de estrategias de protección pulmonar (EPP) para prevenir este tipo de lesión. Una vez instituidas, las EPP han demostrado una disminución de la mortalidad de aproximadamente 10%.
Palabras clave. Daño pulmonar asociado a ventilación mecánica. PEEP. Volumen corriente bajo. Estrategias de protección pulmonar.
INTRODUCCIÓN
Las técnicas de ventilación con presión positiva fueron desarrolladas en la primera mitad del siglo XX para brindar apoyo ventilatorio durante la anestesia de cirugías torácicas, siendo incorporadas para su uso en pacientes críticos a partir de los años 50's, desde entonces la AMV es un soporte indispensable en pacientes críticamente enfermos que tienen como principal problema la insuficiencia respiratoria aguda.
A pesar del indiscutible valor terapéutico de la AMV, su uso está ligado con complicaciones que en algunos casos pudiesen poner en peligro la vida del enfermo, tales como: incremento del riesgo para desarrollar neumonía nosocomial (aproximadamente 20% de posibilidades con sólo iniciar la AMV), alteración de la función cardiaca particularmente en pacientes hipovolémicos, efectos adversos de la sedación y la relajación muscular y recientemente, ha cobrado importancia una de las complicaciones potenciales más serias del uso de la AMV conocida como DPVM, cuyas formas descritas son: barotrauma, volutrauma, atelectrauma, biotrauma y efectos tóxicos causados por oxígeno (Cuadro 1).
DETERMINANTES DE DAÑO PULMONAR ASOCIADO A LA VENTILACIÓN MECÁNICA
Presión en la vía aérea y distensión pulmonar en ventilación espontánea
El pulmón es una estructura elástica que se colapsa y expulsa todo su aire por la tráquea si no existe una fuerza que lo mantenga inflado. No hay un punto de fijación entre el pulmón y las paredes de la caja torácica, excepto en el lugar en el que se encuentra suspendido del mediastino por el hilio.
Para favorecer el flujo del aire hacia el pulmón se requiere de un gradiente de presión entre la atmósfera y el alvéolo. Al final de la espiración, cuando los músculos respiratorios están relajados, el pulmón y la pared torácica ejercen fuerzas opuestas; por un lado el pulmón tiende a disminuir su volumen a través de las fuerzas de recuperación elástica hacia adentro y la pared torácica tiende a aumentar su volumen a través del mecanismo de recuperación elástica hacia afuera, de tal manera que el alvéolo tiende a colapsarse y la pared torácica lo impide. Esta interacción favorece la presencia de una presión inferior a la atmosférica en el espacio pleural. Bajo condiciones normales, durante la inspiración, a través de la contracción muscular, se genera una presión aún más negativa, que favorece directamente la entrada del aire hacia el pulmón. La presión que se genera en el espacio pleural, presente aún cuando los músculos inspiratorios no están contraídos, se conoce como presión pleural (PP). Durante la ventilación espontánea, la PP es de aproximadamente –5 cm H2O al inicio de la inspiración, después se crea una presión hasta un valor medio de aproximadamente –7.5 cm H2O, lo cual permite la entrada de aproximadamente 500 mL de aire a los pulmones.
La presión alveolar (PA) se genera por la presencia de aire en el interior de los alvéolos. Cuando la glotis se encuentra abierta, pero no existe flujo de aire ni hacia adentro ni hacia afuera, la PA se iguala con la atmosférica y se considera como 0 cm H2O (cero relativo porque en realidad corresponde a la presión atmosférica). Para que exista flujo de aire hacia el pulmón, la PA debe disminuir del valor atmosférico. Durante la inspiración normal la PA disminuye aproximadamente a –1 cm H2O, suficiente para favorecer el flujo de 500 mL de aire a los pulmones; durante la espiración sucede lo contrario y se eleva hasta +1 cm H2O.
La diferencia de presión entre la PA y la PP se denomina presión transpulmonar (PTP) y representa a las fuerzas elásticas del pulmón que tienden a colapsarlo en cada grado de expansión (presión de retroceso elástico).
El grado de expansión pulmonar por unidad de incremento en la presión transpulmonar se denomina distensibilidad. La distensibilidad pulmonar total es en promedio de 200 mL de aire por cada cm H2O de PTP, es decir, por cada cm de H2O de incremento en la PTP ingresan 200 mL de aire al pulmón. La curva de distensibilidad pulmonar se ilustra en la figura 1.
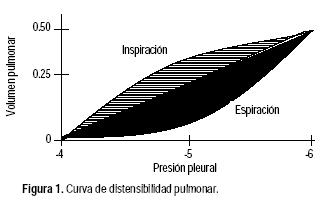
Presión en la vía aérea y distensión pulmonar en ventilación mecánica
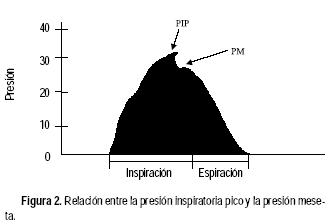
Las presiones durante la AMV son inversas, ya que el principio de la misma se basa en el uso de presión positiva. Durante la AMV, la presión de la vía aérea aumenta progresivamente hasta alcanzar la presión inspiratoria pico (PIP), la cual es resultado de la suma de dos presiones diferentes:
• Aquella que se requiere para vencer la resistencia de la vía aérea y
• La presión necesaria para superar las propiedades elásticas retráctiles del pulmón y de la caja torácica.
Si se agrega una pausa al final de la inspiración (flujo cero con la vía aérea cerrada) la presión cae ligeramente por debajo de la PIP y se denomina presión meseta o plateau (PM), ésta refleja únicamente la presión necesaria para vencer las fuerzas elásticas de la vía aérea distal y se constituye como el mejor estimado de la presión alveolar pico (PAP), un indicador importante de sobredistensión alveolar y por lo tanto, un parámetro de riesgo para complicaciones. Las relaciones entre ambas presiones se ilustran en la figura 2. Las curvas de distensibilidad pulmonar en el enfermo con ventilación mecánica sólo son diferentes por el hecho de manejar presión positiva; sin embargo, están basadas en la misma relación de presiones y volúmenes. En la curva de presión–volumen se observa un patrón sinusoide, donde al principio el volumen ingresa al sistema pulmonar sin ocasionar grandes cambios de presión hasta alcanzar un punto de cambio, denominado punto de inflexión inferior que representa el punto de apertura alveolar, se observa entonces un aumento rápido de la presión con cambios mínimos de volumen hasta el final del esfuerzo inspiratorio, donde se forma el punto de inflexión superior, que marca el área con un mayor número de alvéolos abiertos, para dar paso a la pendiente espiratoria.
En los enfermos con síndrome de insuficiencia respiratoria aguda (SIRA) se presenta un aumento del espacio muerto y reducción de la distensibilidad pulmonar, lo cual contribuye al incremento del trabajo ventilatorio, aunque la principal causa de hipoxemia es el mayor número de cortocircuitos intrapulmonares. Sin la AMV, muchos de estos enfermos desarrollarán (en horas o días) hipoxemia grave, hipercarbia, acidosis y, finalmente, fallecerán. Por mucho tiempo se utilizó libremente un volumen corriente (VC) entre 10–15 mL/kg para cumplir los objetivos de mantener una PaCO2, pH y la PaO2 en rangos próximos a los normales, con niveles bajos12 de FiO2; sin embargo, la ventilación con VC y PIP elevados puede sobredistender las zonas pulmonares teóricamente bien ventiladas. En modelos experimentales con animales, se ha demostrado que la sobredistensión alveolar causa inflamación, aumento de la permeabilidad vascular, pérdida de la función del surfactante entre otras manifestaciones de daño pulmonar agudo, indistinguible del observado en el SIRA.3–7 En animales ventilados con PIP de 14 cm H2O no se encontraron cambios histológicos a nivel pulmonar, pero con niveles de 30 cm H2O, se observa la presencia de edema perivascular leve. En estudios de ventilación mecánica con ratas utilizando PIP de 45 cm H2O sin presión positiva al final de la espiración (PEEP) se observó desarrollo de hipoxia grave, con marcado edema alveolar y perivascular.8
También se ha reportado la presencia de DPVM con el uso de VC y de PEEP relativamente bajos.7,9–11El mecanismo de esta forma de DPVM aún no está claro; sin embargo, un mecanismo propuesto es el estrés excesivo entre las regiones ventiladas y no ventiladas,12 además, la apertura y cierre repetidos de bronquios y alvéolos resultan en fuerzas mecánicas perjudiciales6 con deterioro de la función del surfactante secundario a grandes cambios en el área de superficie alveolar.5,13 Este tipo de DPVM puede ser atenuado con el uso de niveles altos de PEEP o con la aplicación de maniobras de reclutamiento alveolar;11,14–16 sin embargo, se debe tener cuidado con el compromiso hemodinámico asociado con el aumento de la presión intratorácica17 y contribuir al DPVM por sobredistensión.
El incremento en la frecuencia respiratoria puede aumentar el daño pulmonar a través de grandes ciclos de estrés, un fenómeno bien descrito en ingeniería o a través de la desactivación del surfactante. Un estudio en pulmones de conejos ventilados con frecuencias respiratorias de 20 por minuto, muestran mayor edema y hemorragia perivascular que los ventilados con tres respiraciones por minuto,18 estos hallazgos no han sido demostrados en la práctica clínica.
Fracción inspirada de oxígeno (FiO2)
Se considera que el estrés oxidativo es un importante mecanismo de daño pulmonar agudo en pacientes con enfermedades como el SIRA. En humanos y animales la aplicación de FiO2 altas producen daño pulmonar probablemente a través del incremento en la generación de especies reactivas de oxígeno.19,20 En la actualidad, los pacientes con SIRA se manejan con FiO2 baja, tratando de mantener una saturación de oxígeno de por lo menos 90%.
El daño pulmonar no es un problema clínico en pacientes con pulmones sanos, éstos pueden tener apoyo mecánico ventilatorio por tiempo prolongado sin efectos deletéreos.21
MANIFESTACIONES DEL DPVM
Edema pulmonar
El edema pulmonar es el hallazgo fundamental en modelos experimentales de DPVM, particularmente donde se involucran animales pequeños.9,22,23 El alto contenido de proteínas en el líquido de edema sugiere, al menos en parte, que hay un incremento de la permeabilidad. Algunos estudios experimentales demostraron cambios en el epitelio y en la barrera endotelial microvascular. Un estudio en corderos sugiere que la ventilación con volúmenes corrientes altos causa un incremento discreto de la presión transmural vascular pulmonar.24 Las presiones hidrostáticas deben ser tomadas en cuenta, dado que las diferencias regionales en la perfusión pulmonar y las atelectasias pueden generar un gradiente mayor en la fuerza de filtración de algunas áreas,25 así como también el incremento constante en la presión transmural que facilita la salida de líquido de edema hacia el espacio alveolar.
CAMBIOS MORFOLÓGICOS
Los cambios agudos desde el punto de vista estructural en el DPVM se han definido mediante modelos animales. La microscopía de luz demuestra la formación de edema perivascular, el cual progresa hasta edema intersticial y alveolar. Los cambios en el endotelio son notables por microscopía electrónica con sólo pocos minutos de ventilación con presiones altas en la vía aérea en ratones de experimentación, principalmente separación de la membrana basal en algunas células endoteliales y eventualmente es evidente el daño difuso en la superficie alveolar con destrucción de células tipo I y en menor proporción de células tipo II.3,23 En estudios con cerdos ventilados utilizando PIP y PM elevadas, se observó reclutamiento de neutrófilos en 24 horas y cambios fibroproliferativos después de 3–6 días.6
A pesar de todo, el incremento en la permeabilidad vascular, el daño alveolar difuso, los infiltrados celulares inflamatorios y los cambios fibroproliferativos no son específicos del DPVM; como se mencionó previamente, el SIRA y otras formas de daño pulmonar están asociadas con hallazgos histológicos similares.8
El mecanismo fisiopatológico del aire extraalveolar fue descrito por Macklin y Macklin en 1994.26 En su trabajo se establece que durante la ventilación mecánica con presión positiva se rompen alvéolos en el punto en que la base alveolar llega a la vaina bronco–vascular; posteriormente el aire se diseca a lo largo del trayecto de las vainas vasculares hacia el hilio y los tejidos blandos del mediastino; cuando se acumula suficiente gas se rompe la pleura parietal mediastínica y se desarrolla neumotorax. En un estudio con enfermos en AMV se observó barotrauma en 34 casos,27 en relación directa con la PIP, el nivel de la PEEP, el VC y la ventilación por minuto. La relación de la PEEP con la formación de aire ex–traalveolar aún no está clara.28,29 Los pacientes con una enfermedad pulmonar grave concomitante suelen requerir valores más altos de PEEP para mantener una oxigenación adecuada y es probable que la enfermedad pulmonar asociada explique la correlación entre la fuga de aire y los valores de la PEEP. Los enfermos con daño pulmonar (asma bronquial, enfermedad pulmonar obstructiva crónica y neumonía) tienen mayor predisposición al desarrollo de barotrauma. En algunos estudios se ha demostrado una incidencia aproximada de 40% de formación de aire extraalveolar en pacientes con SIRA.27,28,30 El pronóstico empeora en los pacientes que reciben AMV y desarrollan barotrauma.
Webb y Tierney ilustraron gráficamente los efectos nocivos de la ventilación mecánica en ratas utilizando diferentes niveles de presión máxima de la vía aérea y de PEEP.9 Las ratas que se ventilaron con PIP de 14 cm H2O y sin PEEP, no presentaron ninguna alteración histológica ni fisiológica. En cambio las ratas ventiladas con presiones máximas de 30 cm H2O y sin PEEP mostraron edema perivascular sin edema alveolar. Las ratas que recibieron PIP de 45 cm H2O desarrollaron edema alveolar y perivascular, hipoxia grave, disminución de la adaptabilidad dinámica y anormalidades anatómicas.
El aumento del líquido de edema que se produce durante la ventilación mecánica se relaciona con una mayor filtración del mismo hacia el espacio intersticial,31,32 y se ha demostrado que la insuflación progresiva del pulmón involucra a una caída de la presión intersticial33 además de provocar dilatación de los vasos extraalveolares, con el efecto neto de un aumento del gradiente hidrostático para llevar líquido de los capilares al espacio intersticial.33–35 Por otro lado, la ventilación con VC alto puede originar incremento de la permeabilidad de la barrera alveolocapilar a las proteínas. Diferentes estudios han encontrado un mayor coeficiente de filtración durante la ventilación mecánica,4,36 lo cual obedece a una reducción de la integridad de la barrera alveolocapilar y sugiere que durante la ventilación mecánica se lesionan la membrana epitelial y endotelial. En animales de experimentación que se ventilan por cortos periodos de tiempo (dos minutos) se puede desarrollar con rapidez tanto el edema como la lesión de la barrera alveolocapilar con irregularidades del coeficiente de filtración.37 Otros estudios han demostrado que durante la ventilación por periodos más largos se presenta agravamiento progresivo del edema. En ratas ventiladas por espacio de 5–20 minutos se observó un incremento en la acumulación de líquido extravascular, progresando de edema perivascular hasta edema intersticial y edema alveolar. A los 20 minutos se observaron daño de las células tipo I, denudamiento de la membrana basal y formación de membrana hialina. Los animales pequeños y jóvenes parecen más susceptibles a los efectos deletéreos de las presiones altas en la vía aérea, probablemente por inmadurez de la barrera alveolocapilar.3,9,36,38
La distensión excesiva de las unidades alveolares puede conducir a la alteración mecánica de la barrera alveolocapilar y al desarrollo de edema pulmonar.39 La variabilidad de la distribución del aire con diferentes tiempos de apertura alveolar produce cambios regionales en la adaptabilidad del pulmón que puede explicar las diferencias regionales de la insuflación pulmonar y de la distribución del daño.
Otro de los aspectos a considerar en el desarrollo de DPVM es la velocidad del flujo de gas. Peevy et al. demostraron que los flujos altos se acompañan de un aumento del coeficiente de filtración independientemente del nivel de presión inspiratoria utilizada.40
Recientemente se ha prestado considerable atención a la liberación de mediadores inflamatorios del tejido pulmonar expuesto a las fuerzas mecánicas. Algunos estudios han evidenciado que algunas estrategias ventilatorias están asociadas con la liberación de diferentes mediadores proinflamatorios incluyendo el tromboxano B2, el factor activador de plaquetas y diferentes citocinas.11,41–44 La importancia de estos mediadores en la causalidad del daño pulmonar es incierta, aunque existe evidencia que sugiere que este tipo de respuesta inflamatoria está ligada con la perpetuación del daño y al momento actual se le conoce como biotrauma.45 En estudios donde se han utilizado conejos con DPVM, se ha demostrado que el daño pulmonar puede ser atenuado por la administración de anticuerpos antifactor de necrosis tumoral alfa (Ac anti TNFα) o antagonistas de los receptores para interleucina–1 (IL–1), lo cual sugiere que estas citocinas ejercen ciertamente un efecto deletéreo.46,47 El término biotrauma entonces, ha sido designado para describir el potencial deletéreo de la respuesta inflamatoria al estrés físico.
Se ha observado en modelos experimentales en ratas con daño pulmonar que la lesión provocada por la ventilación se asocia a un incremento de los niveles de citocinas en la circulación y algunos estudios recientes sugieren que puede ser aplicado en el escenario clínico.42,43,48,49 Esto ha llevado a plantear la teoría de que la ventilación mecánica desencadena el síndrome de respuesta inflamatoria sistémica (SRIS) habitualmente vista en el SIRA y contribuye al desarrollo de falla orgánica múltiple (FOM).50 La ventilación mecánica también puede desencadenar SRIS a través de la traslocación de bacterias o sus productos del espacio aéreo a la circulación.8
ESTRATEGIAS DE PROTECCIÓN PULMONAR (EPP)
A partir del conocimiento de las condicionantes del DPVM se han desarrollado algunas estrategias de protección pulmonar (EPP) contra los efectos del uso de la presión positiva a través de la AMV.
En los 90's se evaluaron cinco estudios clínicos aleatorizados comparando la estrategia tradicional de AMV vs. el uso de medidas de protección pulmonar (Cuadro 2). En dos de los estudios, la mortalidad fue menor en el grupo que recibió EPP; sin embargo, en los otros tres no se observó beneficio e inclusive los datos arrojaron cierta tendencia al aumento en la mortalidad. Es importante mencionar que existen diferencias importantes en cuanto a la metodología utilizada en los diferentes estudios, entre ellas el nivel de VC utilizado, el poder del estudio, el uso de otras EPP y el manejo de la acidosis; lo cierto es que entre mayor era la diferencia en el VC mejor era el pronóstico, inclusive la proporción de enfermos que desarrollaron DPVM fue mayor en tanto el VC utilizado era más alto, particularmente con los niveles utilizados en el grupo de enfermos tratados con la estrategia convencional (VC 10–15 mL/kg).
Amato et al.51 reportaron resultados alentadores mediante la reducción de la mortalidad utilizando AMV con VC de aproximadamente 6 mL/kg (en este grupo de enfermos, el VC fue disminuido inclusive por debajo de 5 mL/kg si la presión en la vía aérea excedía los 40 cm H2O), y una PEEP promedio de 16.4 cm H2O durante las primeras 36 horas comparado con la estrategia convencional utilizando VC de 12 mL/ kg y una PEEP promedio de 8 cm H2O durante los primeros siete días. En el grupo manejado con EPP se utilizaron maniobras de reclutamiento alveolar que incrementaron la presión en la vía aérea hasta 35–40 cm H2O durante 40 segundos para el manejo de atelectasias y para prevenir el daño que se produce por la apertura y cierre repetidos de los bronquiolos respiratorios y de las unidades alveolares, además de atenuar el estrés excesivo sobre los márgenes entre las unidades pulmonares ventiladas y no ventiladas. En el grupo de pacientes sometidos a las EPP se observó aumento en la sobrevida y en el porcentaje de liberación de la AMV, mientras que la frecuencia de eventos de barotrauma fue menor. Sin embargo, no está claro si los resultados clínicos pueden ser atribuidos solamente al uso de VC bajo, de PIP limitada, niveles altos de PEEP, maniobras de reclutamiento o a la combinación de estos elementos. Una de las limitantes del estudio es el pequeño tamaño de la muestra, con un grupo de 24 pacientes en el grupo con medidas convencionales y 29 pacientes en el grupo de EPP.
En el estudio de los NIH (The National Institutes of Health) se observó disminución de la mortalidad de 40 a 31%, evaluando únicamente el uso de VC bajo como EPP, con 861 enfermos; mientras que la combinación de los otros tres estudios que también evaluaron únicamente el VC como EPP con resultados negativos, fue de 288 enfermos. Otra de las diferencias en el protocolo utilizado por los NIH fue el manejo más estricto de la acidosis respiratoria, que incluía aumento de la frecuencia respiratoria y el uso de bicarbonato con el objetivo de mantener un pH por arriba de 7.3, situación que pudiese haber influido en el desenlace de estos enfermos.
De los resultados obtenidos en el protocolo del NIH, se advierte claramente que el uso de EPP con VC bajo favorece un mejor pronóstico; sin embargo, es necesario considerar que los resultados son aplicables únicamente a los enfermos con SIRA.56
Las EPP recomendadas para su uso de forma temprana durante el curso del SIRA con la finalidad de evitar el desarrollo de DPVM, se describen en el cuadro 3.
CONCLUSIÓN
La ventilación mecánica está asociada con grandes beneficios clínicos en los enfermos con insuficiencia respiratoria, sin embargo, no es del todo inocua, de hecho se asocia con complicaciones que en algunos casos puede poner en peligro la vida del enfermo. El DPVM es una de las complicaciones potenciales más graves; las variantes que comprende son: barotrauma, volutrauma, atelectrauma, biotrauma y efectos tóxicos causados por oxígeno. Se ha propuesto que el mecanismo principal está relacionado con la presencia de sobredistensión alveolar, lo cual conduce a diversos efectos tanto en la pared alveolar como en la pared vascular, produciendo aumento en la concentración de citocinas inflamatorias, aumento de la permeabilidad vascular y edema pulmonar rico en proteínas. Debido a la estrecha relación entre la sobredistensión y daño, se han propuesto estrategias de protección pulmonar, todas encaminadas a disminuir la presión meseta (plateau) e indirectamente la presión alveolar pico. A pesar de la controversia de los resultados, se acepta que con estas medidas se ha logrado disminuir la mortalidad hasta 10% aproximadamente. Esto resalta la importancia del monitoreo continuo de las medidas de distensibilidad pulmonar y la intervención temprana en situaciones de riesgo.
REFERENCIAS
1. Colice GL. Historical perspective on the development of mechanical ventilation. In: Principles and Practice of Mechanical Ventilation. Tobin MJ (Ed.). New York: McGraw– Hill; 1994, p. 1–35. [ Links ]
2. Marini JJ. Evolving concepts in ventilatory management of acute respiratory distress syndrome. Clin Chest Med 1996; 17: 555–75. [ Links ]
3. Dreyfuss D, Soler P, Basset G, et al. High inflation pressure pulmonary edema. Am Rev Respir Dis 1988; 137: 1159–64. [ Links ]
4. Parker JC, Townsley MI, Rippe B, et al. Increased microvascular permeability in dog lungs due to high peak airway pressures. J Appl Physiol 1984; 57: 1809–16. [ Links ]
5. Greenfield LJ, Ebert PA, Benson DW. Effect of positive pressure ventilation on surface tension properties of lung extracts. Anesthesiology 1964; 25: 312–16. [ Links ]
6. Tsuno K, Miura K, Taakeya M, et al. Histopathologic pulmonary changes from mechanical ventilation at high peak airway pressure. Am Rev Respir Dis 1991; 143: 1115–20. [ Links ]
7. Corbridge TC, Wood LDH, Crawford GP, et al. Adverse effect of large tidal and low PEEP in canine acid aspiration. Am Rev Respir Dis 1990; 142: 311–15. [ Links ]
8. Whitehead T, Slutsky AS. The pulmonary physician in critical care 7: Ventilator–induced lung injury. Thorax 2002; 57: 635–42. [ Links ]
9. Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high pressure. Am Rev Respir Dis 1974; 110: 556. [ Links ]
10. Muscedere JG, Mullen JBM, Gan K, et al. Tidal ventilation at low airway pressure can augment lung injury. Am J Respir Crit Care Med 1994; 149: 1327–34. [ Links ]
11. Tremblay L, Valenza F, Ribeiro SP, et al. Injurious ventilatory strategies increase cytokines and c–fos–m–RNA expression in an isolated rat lung model. J Clin Invest 1997; 99: 944–52. [ Links ]
12. Mead J, Takishima T, Leith D. Stress distribution in lung: A model of pulmonary elasticity. J Appl Physiol 1970; 28: 596–608. [ Links ]
13. Faridy EE, Permutt S, Riley RL. Effect of ventilation on surface forces in excised dog's lungs. J Appl Physiol 1996; 21: 1453–62. [ Links ]
14. Lachman B. Open up the lung and keep the lung open. Intensive Care Med 1992; 18: 319–21. [ Links ]
15. Rimensberger PC, Cox PN, Frndova H, et al. The open lung during small tidal volume ventilation: Concepts of recruitment and "optimal" positive end–expiratory pressure. Crit Care Med 1999; 27: 1946–52. [ Links ]
16. Marini JJJ, Amato MBP. Lung recruitment during ARDS. In: Lung injury. Marini JJ, Evans TW (Ed.). Berlin: Springer; 1998, p. 236–57. [ Links ]
17. Pinsky MR. Heart–lung interactions. In: Pathophysiologic Foundations of Critical Care. Pinsky MR, Dhainaut JF (Ed.). Baltimore: Williams & Wilkins; 1993, p. 472–90. [ Links ]
18. Hotchkiss JR, Blanch L, Murías G, et al. Effects of decreased respiratory frequency on ventilator–induced lung injury. Am J Respir Crit Care Med 2000; 161: 463–8. [ Links ]
19. Chabot F, Mitchell JA, Gutteridge JM, et al. Reactive oxygen species in acute lung injury. Eur Respir J 1998; 11: 745–57. [ Links ]
20. Davis WB, Rennard SI, Bitterman PB, et al. Pulmonary oxygen toxicity. Early reversible changes in human alveolar structures induced by hyperoxia. N Engl J Med 1983; 309: 878–83. [ Links ]
21. Nash G, Bowen JA, Langlinais PC. "Respirator lung": A misnomer. Arch Pathol 1971; 91: 234–40. [ Links ]
22. Kolobow T, Moretti MP, Fumagalli R, et al. Severe impairment in lung function induced by high peak airway pressure during mechanical ventilation. An experimental study. Am Rev Respir Dis 1987; 135: 312–5. [ Links ]
23. Dreyfuss D, Saumon G. Ventilator–induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 1998; 157: 294–323. [ Links ]
24. Carlton DP, Cummings JJ, Scheerer RG. Lung overexpansion increases pulmonary microvascular protein permeability in young lambs. J Appl Physiol 1990; 69: 577–83. [ Links ]
25. Albert RK, Lakshminarayan S, Kirk W, et al. Lung inflation can cause pulmonary edema in zone I of in situ dog lungs. J Appl Physiol 1980; 49: 815–9. [ Links ]
26. Mackiln MT, Macklin CC. Malignant interstitial emphysema of the lungs and mediastinum as an important occult complication in many respiratory diseases and other conditions: An interpretation of the clinical literature in the light of laboratory experiments. Medicine 1944; 23: 281. [ Links ]
27. Gammon BR, Shin MS, Buchalter SE. Pulmonary barotrauma in mechanical ventilation. Patterns and risk factors. Chest 1992; 102: 568. [ Links ]
28. Gammon RB, Shin MS, Groves RH Jr, et al. Clinical risk factors for pulmonary barotrauma: A multivariate analysis. Am J Respir Crit Care Med 1995; 152: 1235. [ Links ]
29. Petersen GW, Baier H. Incidence of pulmonary barotrauma in a medical ICU. Crit Care Med 1983; 11: 67. [ Links ]
30. Woodring JH. Pulmonary interstitial emphysema in the adult respiratory distress syndrome. Crit Care Med 1985; 13: 786. [ Links ]
31. Parker JC, Hernandez LA, Longenecker GL, et al. Lung edema caused by high peak inspiratory pressure in dogs. Role of increase microvascular filtration pressure and permeability. Am Rev Respir Dis 1990; 142; 321. [ Links ]
32. Parker JC, Hernandez LA, Peevy KJ. Mechanism of ventilator–induced lung injury. Crit Care Med 1993; 21: 131. [ Links ]
33. Dreyfuss D, Saumon G. Ventilator–induced lung injury. In: Principles and Practice of Mechanical Ventilation. Tobin MJ (Ed.). New York: McGraw–Hill; 1994, p. 793. [ Links ]
34. Albert RK, Lakshminarayan WK, Butler J. Lung inflation can cause pulmonary edema in zone I of in situ dog lungs. J Appl Physiol 1980; 49: 815. [ Links ]
35. Smith JC, Mitzner W. Analysis of pulmonary vascular interdependence in excised dog lobes. J Appl Physiol 1980; 48: 450. [ Links ]
36. Egan E. Lungs inflation, lung solute permeability and alveolar edema. J Appl Physiol 1982; 53: 121. [ Links ]
37. Dreyfuss D, Soler P, Saumon G. Spontaneous resolution of pulmonary edema caused by short periods of cyclic overinflation. J Appl Physiol 1992; 72: 2081. [ Links ]
38. Adkins KW, Hernandez LA, Coker PJ, et al. Age affects susceptibility to pulmonary barotrauma in rabbit. Crit Care Med 1991; 19: 390. [ Links ]
39. Fu Z, Costello ML, Tsukimoto K, et al. High lung volume increases stress failure in pulmonary capillaries. J Appl Physiol 1992; 73: 123. [ Links ]
40. Peevy KJ, Hernandez LA, Moise AA, Parker JC. Barotrauma and microvascular injury in lungs of nonadult rabbits: Effect of ventilation pattern. Crit Care Med 1990; 18: 634. [ Links ]
41. Imai Y, Kawano T, Miyasaka K, et al. Inflammatory chemical mediators during conventional ventilation and during high frequency oscillatory ventilation. Am J Respir Crit Care Med 1994; 150: 1550–4. [ Links ]
42. von Bethmann AN, Brasch F, Nusing R, et al. Hyperventilation induces release of cytokines from perfused mouse lung. Am J Respir Crit Care Med 1998; 157: 263–72. [ Links ]
43. Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med 1999; 160: 109–16. [ Links ]
44. Held H, Boettcher S, Hamann L, et al. Ventilation–induced chemokine and cytokine release is associated with activation of NF[kappa]B and is blocked by steroids. Am J Respir Crit Care Med 2001; 163: 711–6. [ Links ]
45. Plotz FB, van Vught H, Heijnen CJ. Ventilator–induced lung inflammation: is it always harmful? Intensive Care Med 1999; 25: 236. [ Links ]
46. Imai Y, Kawano T, Iwamoto S, et al. Intratracheal antitumor necrosis factor–alpha antibody attenuates ventilator–induced lung injury in rabbits. J Appl Physiol 1999; 87: 510–5. [ Links ]
47. Narimanbekov IO, Rozycki HJ. Effect of IL–1 blockade on inflammatory manifestations of acute ventilator–induced lung injury in a rabbit model. Exp Lung Res 1995; 21: 239–54. [ Links ]
48. Haitsma JJ, Uhlig S, Goggel R, et al. Ventilator–induced lung injury leads to loss of alveolar and systemic compartmentalization of tumor necrosis factor–alpha. Intensive Care Med 2000; 26: 1515–22. [ Links ]
49. Ranieri V, Suter P, Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 1999; 282: 54–61. [ Links ]
50. Slutsky AS, Tremblay LN. Multiple system organ failure. Is mechanical ventilation a contributing factor? Am J Respir Crit Care Med 1998; 157: 1721–5. [ Links ]
51. Amato MBP, Barbas CSV, Medeiros DM, et al. Effect of a prospective–ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 1998; 338: 347–54. [ Links ]
52. Acute Respiratory Distress Syndrome Network; ventilation with lower tidal for acute lung injury and the acute respiratory distress syndrome. N Engl Med 2000; 342: 1301–8. [ Links ]
53. Brochard L, Roudot–Thoraval F, Roupie E, et al. Tidal volume reduction for prevention of ventilator–induced lungs injury in the acute respiratory distress syndrome. Am J Respir Crit Care Med 1998; 158: 1831–8. [ Links ]
54. Stewart TE, Meade MO, Cook DJ, et al. Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. N Engl J Med 1998; 338: 355–61. [ Links ]
55. Brower RG, Shanhotlz CB, Fessler HE, et al. Prospective randomized, controlled clinical trial comparing traditional vs. reduced tidal volume ventilation in ARDS patients. Crit Care Med 1999; 27: 1492–8. [ Links ]
56. Brower RG, Rubenfield GD. Lung–protective ventilation strategies in acute lung injury. Crit Care Med 2003; 31: S312–S316. [ Links ]
57. Roupie E, Dambrosio M, Servillo G, et al. Titration of tidal volume and induced hypercapnia in acute respiratory distress syndrome. Am J Respir Crit Care Med 1995; 152: 121. [ Links ]

