










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista de investigación clínica
versão On-line ISSN 2564-8896versão impressa ISSN 0034-8376
Rev. invest. clín. vol.57 no.3 Ciudad de México Mai./Jun. 2005
Artículo original
Análisis de expresión global del cáncer cérvico uterino: rutas metabólicas y genes alterados
Global expression analysis in uterine cervical cancer: Metabolic pathways and altered genes
Guelaguetza Vázquez–Ortíz,* Patricia Piña–Sanchez,* Alfredo Hidalgo,*** Minerva Lazos,*** José Moreno,**** Isabel Alvarado,** Fernando cruz,***** Dulce M. Hernández,* Carlos Pérez–Plascencia,* Mauricio Salcedo*
* Unidad de Investigación Médica en Enfermedades Oncológicas,
** Departamento de Patología Hospital de Oncología, Centro Médico Nacional Siglo XXI.
*** Departamento de Patología,
**** Centro Nacional de Clínica de Displasias. Hospital General de México, SS.
***** Unidad de Investigación Médica en Enfermedades Autoinmunes, Hospital de Especialidades, CMN SXXI–IMSS.
Reimpresos:
Dr. Mauricio Salcedo
Unidad de Investigación Médica en Enfermedades Oncológicas.
Hospital de Oncología
Centro Médico Nacional Siglo XXI–IMSS
Av. Cuauhtemoc 330. Col Doctores.
06720, México, D.F.
Phone: +55 56276900 Ext: 22708 Fax: +55 57610952
Email: maosal89@ yahoo.com, guelav@yahoo.com
Recibido el 6 de septiembre de 2004.
Aceptado el 20 de enero de 2005.
ABSTRACT
High risk human papillomavirus (HPV) infection is considered to be the most important etiological factor of Cervical Uterine Cancer. In order to determine the global expression pattern and to identify possible molecular markers of cervical cancer, cDNA arrays with probe sets complementary to 8,000 human genes were used to examine the expression profiles among 5 cell lines derived from human cervical cancer, three HPV16(+) tumor samples and three normal cervical tissues HPV(–). The levels of expression of different cellular processes were identified. Hierarchical clustering was performed and the gene expression using RT–PCR was confirmed. Two genes were found to be consistently overexpressed in invasive cervical cancer biopsies; one of them, IL–6 was previously reported to be overexpressed in cervical cancer and one novel gene, MMP10, previously not known to be related to cervical cancer. Hierarchical clustering of the expression data revealed that samples with common HPV type infection grouped together, maybe this could mean that differences between HPV types could be indirectly determined by expression profiles.
Key words. HPV. Uterine cervical cancer. Microarrays. Expression.
RESUMEN
La infección por virus de papiloma de alto riesgo (VPH) es considerada como el factor etiológico más importante del cáncer cérvico uterino (CaCU). Con el fin de determinar el patrón de expresión global e identificar algunos posibles genes marcadores del CaCU, se utilizaron microhileras de DNA que contenían 8,000 secuencias que codificaban para transcritos diferentes, para estudiar los perfiles de expresión de cinco líneas celulares derivadas de CaCU, tres muestras tumorales conteniendo VPH 16 y tres muestras normales negativas para la presencia de VPH. Se identificaron los niveles de expresión de genes relacionados con diferentes rutas metabólicas. Se llevó a cabo el análisis de agrupamiento jerárquico y posteriormente se confirmó la sobrexpresión de dos genes mediante RT–PCR. Estos dos genes se encontraron sobrexpresados en biopsias tumorales cervicales. Uno de ellos, el gen de IL6, que ha sido previamente reportado en relación con CaCU, así como el gen de la matriz–metaloproteasa 10 (MMP10) por primera vez relacionado con esta neoplasia. El análisis de agrupamiento jerárquico, además, reveló que las muestras que contienen el mismo tipo viral están asociadas, sugiriendo posibles diferencias en expresión entre tipos virales.
Palabras clave. VPH. Cáncer cérvico uterino. Microhileras. Expresión.
INTRODUCCIÓN
El carcinoma cérvico uterino (CaCU), representa la segunda causa de mortalidad en población femenina a nivel mundial,1 y es una las principales causas de muerte en la población femenina mexicana. Este tumor representa más de 24% de todas las neoplasias malignas reportadas en México, haciendo de esta neoplasia uno de los problemas de salud más graves en México.2
En diferentes reportes de tipo epidemiológico, se han determinado diversos factores de riesgo asociados al desarrollo del CaCU, como el número de parejas sexuales, inicio temprano de vida sexual activa, multiparidad, etc.3,4 Sin embargo, el factor etiológico más importante para el desarrollo del CaCU es la infección con virus de papiloma humano de alto riesgo (VPH–AR)5–8 principalmente los tipos 16 y 18.9
Se ha demostrado que los genomas de los VPH–AR usualmente se integran al genoma de la célula hospedera. Esta integración se realiza en diversos sitios, algunos de ellos cercanos a oncogenes específicos o sitios frágiles, sugiriendo que este evento puede tener un papel importante en el desarrollo de los tumores cervicales.10
La propiedad oncogénica principal de las proteínas virales E6 y E7 es su capacidad de inactivar a las proteínas celulares supresoras de tumor pRB y p53,11,12 respectivamente, permitiendo así la transformación e inmortalización de las células infectadas. La inactivación de los genes supresores de tumor, ya sea por mutaciones génicas o por presencia de virus genera la desregulación del ciclo celular y la activación de varios protooncogenes.13 Estas interacciones no han sido totalmente estudiadas14 y se desconoce, por lo tanto, el comportamiento global del CaCU.
En el presente trabajo fueron utilizadas microhileras de expresión (DNAc) para explorar el comportamiento global de la expresión génica en el CaCU. Los patrones de expresión de las diferentes rutas metabólicas estudiadas permiten observar que éstas en general, se encuentran modificadas en el proceso de carcinogénesis cervical con respecto a su contraparte normal.
MATERIAL Y MÉTODOS
Las líneas celulares HeLa (VPH18), SiHa (VPH16) y CaSki (VPH16) se crecieron en el medio de cultivo de Dulbecco. Por otro lado, es importante mencionar que aunque el HPV18 ha sido asociado preferencialmente con adenocarcinomas, también ha sido encontrado en carcinomas escamosos del cervix.15 Otras líneas celulares utilizadas en este trabajo: CALO e INBL que presentan HPV18 derivan de: un carcinoma escamoso invasor del cervix estadio IIB y de un carcinoma escamoso invasor estadio IVA, respectivamente.16 En el presente trabajo se obtuvieron ocho tejidos posmortem del cervix uterino, sin lesión, de mujeres premenopáusicas, cuya muerte no estuvo relacionada con procesos ginecológicos, y sin presencia de miomatosis uterina, del Departamento de Patología del Hospital de Oncología, CMN Siglo XXI. Los tejidos invasores (ocho muestras) fueron obtenidos a través de biopsias guiadas por colposcopia de la lesión tumoral de mujeres premenopáusicas en el Centro Nacional de Displasias del Hospital General de México, S.S. Todos los procedimientos descritos han sido evaluados y aprobados por el comité local de ética del Hospital de Oncología del Instituto Mexicano del Seguro Social y las muestras invasoras fueron tomadas bajo el consentimiento de las pacientes previamente informadas. Todas las muestras se dividieron en tres secciones, la parte media se congeló en nitrógeno líquido y se almacenó a –70 °C hasta su uso. Los extremos de la biopsia fueron fijados en etanol al 70% e incluidos en parafina. A diferentes cortes histológicos de los extremos de los tejidos fijados y embebidos en parafina se les realizó la tinción de hematoxilina/eosina con el fin de confirmar la presencia de al menos 80% de células tumorales presentes en la biopsia. Mediante estos cortes, también se realizó la clasificación del tumor. Todas las muestras invasoras fueron diagnosticadas como lesiones escamosas.
Tanto el DNA como el RNA fueron extraídos de las líneas celulares, así como de los tejidos congelados con trizol (Life Technologies, Grand Island NY USA). Con el fin de determinar el tipo viral presente en las muestras o bien la ausencia del virus en las muestras normales, el DNA de cada una de éstas fue sometido a reacción en cadena de la polimerasa o PCR utilizando los oligonucleótidos consenso MY09/ MY11 y secuenciación automatizada de DNA.
El RNA fue tratado con RNAse–free DNAse I (Ambion Inc). La concentración de RNA total fue determinada por absorbancia y la integridad del ácido nucleico fue confirmada por electroforesis. El RNA mensajero se aisló de HeLa, SiHa, CaSki, CaLO, INBL, de tres muestras normales (N03, Nil, N22) y de tres muestras tumorales VPH16 (T07, T31 y T64) mediante el kit Polyatract Isolation System II (Promega Madison, WI), desafortunadamente del resto de muestras tumorales y normales no se obtuvo la cantidad de mensajero necesaria; de ahí que éstas fueran utilizadas para realizar RT–PCR. Sin embargo, es importante mencionar que en la actualidad aún no existe publicado ningún método estadístico que permita definir un tamaño muestral específico para un análisis por microarrays; y el número de muestras que utilizamos en este manuscrito es similar al que se utilizan en diversos artículos publicados en donde usan la misma tecnología. Los RNAs mensajeros se cuantificaron utilizando el VersaFluor Fluorometer (BioRad). Los DNAc marcados fueron obtenidos por la reacción de transcripción reversa acoplada a PCR o RT–PCR a partir de 800 ng de ARNm en presencia de (α–33P) dATP utilizando el kit cDNA Labeling Ambion Advantage System (Ambion Inc, USA). Se realizó la hibridación de las microhileras que contienen 8,400 clonas por duplicado (ULTRArray Advantage System, Arrays and Hibridization, Ambion Inc, Austin Tx) siguiendo las instrucciones del fabricante.
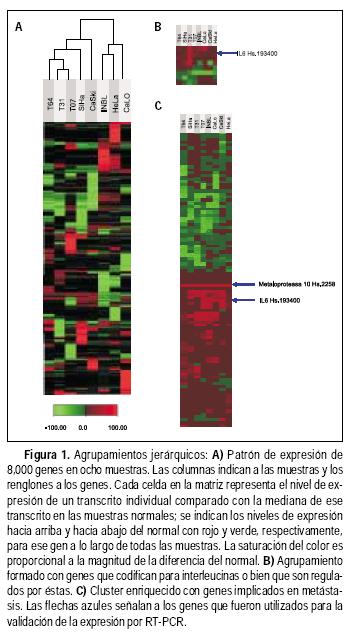
La imagen de las microhileras de DNAc se capturó digitalmente mediante el STORM 860 phosphorimager/fluorimager (Molecular Dynamics, Buckinghamshire UK). Una vez capturadas las imágenes, éstas fueron analizadas y normalizadas con el paquete ArrayVision Software 8.0 rev 4 y LOESS (Imaging Research Inc, USA). Se realizó un corte asignado en los datos, en donde aquellos datos que tuvieran un nivel de expresión menor a 0.5, no fueran tomados en cuenta como sobrexpresados. Las muestras fueron normalizadas contra la mediana de las muestras control (tres muestras normales VPH(–)). La medición de cada gen de las muestras experimentales se hizo dividiendo el valor de muestra entre la mediana de ese gen en su muestra control correspondiente. Con el fin de visualizar los niveles de expresión de los genes en las diferentes muestras, se realizó un agrupamiento o cluster jerárquico utilizando el programa J–Express disponible de manera gratuita en la red (www.molmine.com) y el dendrograma se visualizó con el programa Treeview.
Con el fin de entender el comportamiento biológico del CaCU, se utilizó la base de datos Gene Ontology (http://www.ebi.ac.uk/GOA/) con el fin de clasificar a los genes en diferentes rutas metabólicas. Cada gen presente en el microarreglo fue asociado con su correspondiente número de Unigene (http:// www.ncbi.nlm.nih.gov), y tanto los datos de expresión como los números de Unigene se integraron en el website de Gene Ontology (http://www.geneontology.org) y se procedió a realizar el análisis. Finalmente se obtuvo para cada uno de los genes el Código de Gene Ontology para procesos biológicos, es decir, se obtuvieron las anotaciones funcionales de los genes que componen el arreglo. El programa de Hierarchical clustering (Gene Cluster v2.11) y el programa de visualización (Tree View vl.50) fueron utilizados para graficar los análisis de cada una de las rutas (http://rana.Ibl.gov/Eisensoftware.htm) (Figura 1). Posteriormente se realizó un análisis de medianas. Este se hizo de la siguiente manera: de cada gen a estudiar se obtuvo la mediana de las muestras tumorales y de las líneas celulares y ésta se comparó contra la mediana de las muestras normales VPH(–). Se hizo una sumatoria del total de las medianas de los genes que componen cada ruta metabólica estudiada y así se obtuvo el comportamiento global de la expresión en estas mismas.
Para obtener los DNAc; 200 ng de RNA total de cinco tejidos normales (obtenidos posmortem), las líneas celulares y de cinco muestras tumorales (obtenidas del Centro Nacional de Displasias del Hospital General de México, S.S.) fueron sometidas al uso del estuche de RT–PCR Access System (Promega, Madison, WI). Los oligonucleótidos para los genes de interés fueron diseñados utilizando el programa Biotools de Integrated DNA Technologies (http://biotools.idtdna.com/gateway). Para la amplificación de los siguientes transcritos: Matriz metaloproteasa 10 (MMP10), 509 pb TM = 60 °C (5'–CATTCAGTCTC–TCTACGGACCT–3'), (5'–CAGCTATTAGTCTAGGGA–AGCC–3'). Interleucina 6 (IL6), 621 pb: TM = 55 °C (5'AGTTGCCTTCTCCCTGG3') (5TGAGGGCTCTT–GGGCAAAT–3'), y (3–actin (5'–TGAAGTCTGACGT–GGACATC–3') (GTTCGTTCCTCATACTGCTCA–3') 243 pb, TM = 55 °C. Las condiciones de la RT–PCR fueron las siguientes: 48 °C durante 45 min y 94 °C por dos min. Seguido por 94 °C por 30 seg (la TM °C específica para cada par de oligonucleótidos) durante un min, y 68 °C por dos min en un total de 25 ciclos seguido por un último paso de 68 °C durante siete min. Los productos se visualizaron en geles de garosa al 1.5% teñidos con bromuro de etidio, y las imágenes se capturaron utilizando Eagle Eye System (Stratagene, La Jolla, Ca). Los tamaños de los productos fueron determinados por comparación con un marcador de 100 pb (GeneRuler, MBI Fermentas).
RESULTADOS
Con el fin de estudiar el transcriptoma del cáncer cervical, en el presente trabajo iniciamos con la definición de los patrones de expresión de líneas celulares derivadas de CaCU y de tres muestras de CaCU positivas para VPH16, comparando las medianas de los valores de expresión de las líneas celulares y tejidos enfermos con la mediana de los valores de expresión del cervix normal negativo para secuencias de VPH. De este análisis, se lograron agrupar los genes estudiados en genes sobrexpresados, suprimidos y aquellos que no tuvieron cambios en los valores de expresión con respecto a tejidos normales.
Con el fin de identificar patrones de expresión génica que se modificaran en las muestras utilizadas, se realizó de primera instancia un análisis de tipo de agrupamiento jerárquico con una matriz de visualización seudocoloreada.17 En dicho agrupamiento o cluster jerárquico, el eje "Y" agrupa genes con niveles de expresión similar y en el eje "X" agrupa muestras con expresión similar. Por otro lado, la matriz coloreada del "cluster" permite que la sobreexpresión génica sea fácilmente identificada cuando se comparte entre líneas celulares y muestras (puede visualizarse como un renglón coloreado en rojo). Esto significa que estos genes pueden ser potencialmente utilizados como marcadores. Una propiedad notable de los datos estudiados fue que las muestras y tumores infectados con el mismo tipo de VPH se agrupan en dos ramas distintas en el dendrograma (Figura 1a): es decir, que las líneas celulares SiHa y CaSki (VPH 16) y las muestras tumorales VPH16(+) T07, T31 y T64, así como las líneas celulares VPH18(+) (INBL, HeLa, CaLO) fueron segregadas en dos ramas terminales independientes en el cluster.
Por otro lado, se identificaron grupos de genes involucrados en procesos celulares específicos, la variación en sus niveles de expresión podría reflejar las diferencias en la actividad de estos procesos en las distintas muestras estudiadas. Por ejemplo, en la figura 1b todos los genes identificados pertenecen a los de las interleucinas. En otro cluster no mostrado, las muestras estudiadas reflejan una sobrexpresión de los genes característicos de células epiteliales, no así en los genes pertenecientes a estroma; haciendo notar que las células estudiadas son de origen epitelial y que las biopsias están compuestas en su mayoría por células epiteliales.18 Así, este cluster está constituido por componentes complejos adherentes, moléculas de adhesión célula–célula, e intercambiadores de sodio.19
En la figura le se muestra un cluster específico para genes involucrados en metástasis, como las metaloproteasas, las integrinas involucradas en la arquitectura celular, así como inhibidores de proteasas.
Debido a que uno de los propósitos de este trabajo era encontrar genes que pudieran estar asociados al CaCU, los 15 genes que estuvieran mayormente sobrexpresados en todas las muestras y los 15 genes suprimidos en todas las muestras se seleccionaron para elaborar una tabla basada en aquellos genes con sobrexpresión o subexpresión > 4 veces con respecto al normal (Cuadro 1.)
Expresión de procesos celulares
Con el fin de estudiar la variación de los niveles de RNAm en diferentes rutas metabólicas, utilizamos la base de datos de Gene Ontology para identificar grupos de genes presentes en nuestras microhileras que estuvieran involucrados en procesos celulares específicos. Estas variaciones se presentan en una gráfica de barras (Figura 2). En general puede observarse que todos los procesos celulares mostrados se encuentran preferencialmente sobrexpresados, siendo los más afectados los procesos de ciclo celular y los pertenecientes al grupo de metabolismo de fármacos. Cabe mencionar que la línea celular HeLa muestra un patrón de sobrexpresión muy diferente a las otras líneas celulares con VPH 18. El resto de las rutas presenta cambios muy discretos.
Confirmación de la sobrexpresión de genes en CaCU por RT–PCR
Para validar la expresión diferencial del gen IL6, previamente identificado como posible marcador de CaCU y MMP10 como un nuevo posible gen marcador, ambos obtenidos a partir del cluster jerárquico, se utilizaron dos diferentes métodos de análisis. El primer método utilizado fue la base de datos " Gene to Tag" que realiza la imagen del experimento de tipo northern blot virtual (http://www.ncbi.nlm.nih.gov/Unigene/). En esta aplicación se hibridaron virtualmente cada uno de los dos transcritos estudiados (IL6 y MMP10) contra todas las librerías de expresión de líneas celulares y tejidos existentes en la base de datos. De manera interesante, los dos genes hibridaron y mostraron sobrexpresión en los tejidos de carcinoma de colon, adenocarcinoma de estómago y carcinoma de mama (resultados no mostrados). Con la información obtenida del experimento anterior se procedió a realizar experimentos de RT–PCR en las células HeLa, CaLO, INBL, SiHa, CaSki, en cinco tejidos normales VPH(–) y en cinco biopsias tumorales del cervix. Los resultados de estos experimentos mostraron que hay sobrexpresión de las líneas celulares y de las muestras tumorales con respecto al epitelio normal (Figura 3).
DISCUSIÓN
Con el fin de identificar marcadores potenciales y alteraciones moleculares particulares del cáncer cérvico uterino, utilizamos microhileras de DNAc para determinar los niveles de expresión de más de 8,000 genes en cinco líneas celulares derivadas de CaCU, tres biopsias de CaCU positivos para VHP 16 y tres tejidos cervicales normales negativos para VPH.
Como se esperaba, el dendrograma generado por el agrupamiento jerárquico y los niveles de expresión génica de las muestras, dieron como resultado la identificación de dos subgrupos de genes correspondientes al tipo viral presente en las muestras. Otros genes identificados como sobrexpresados (datos no mostrados) son la inosina monofosfato deshidrogenasa, el factor de necrosis tumoral (TNF) y la metiltransferasa 1. Éstos sólo se encontraron sobrexpresados en las muestras con VPH 16, por lo que es posible que la expresión de éstos y otros genes adicionales permitiera determinar las diferencias específicas entre los tipos virales 16 y 18. Sin embargo, estos resultados son aún preliminares y están pendientes estudios adicionales que permitan establecer su significado funcional.
Con el fin de conocer las diferencias en expresión de procesos celulares específicos de las muestras estudiadas, se identificaron algunos grupos conformados por genes característicos de rutas metabólicas y utilizando una gráfica de barras se mostraron los niveles de expresión (sobrexpresión o supresión) de las muestras estudiadas con respecto a su contraparte normal, este análisis se logró utilizando la base de Gene Ontology. El Gene Ontology Consortium (GO) es un proyecto internacional cuyos resultados están accesibles en la red. Podemos mencionar entre otros aspectos que la base de datos GO muestra un grupo de términos bien definidos y relaciones entre ellos, con lo cual podemos interpretar el papel de un gen en particular, producto de un gen o un grupo de productos de genes involucrados en la patogénesis. El análisis GO organiza a los genes en tres ontologías separadas que comprenden procesos biológicos, componentes celulares y funciones moleculares. Uno de los factores más importantes es que agrupa diversas bases de datos. Su estructura permite que los genes tengan asignadas propiedades en diferentes niveles, dependiendo de cuánto se sabe sobre el producto de ese gen. De ahí que actualmente sea una de las herramientas más importantes para el análisis de datos obtenidos por microhileras.20
El grupo de genes propios de células epiteliales está conformado por: periplakina, desmoplakina, junction plakoglobina, y claudina 4, entre otros; no presenta cambios en los niveles de expresión en las diferentes muestras, sugiriendo que estos transcritos se originaron en células tumorales con características epiteliales similares. El grupo de genes de proliferación y de ciclo celular básicamente están compuestos por productos que son necesarios para la progresión a través del ciclo celular, para procesamiento de RNA y maquinaria de traducción y marcadores para identificar células en proliferación.21 En el grupo de proliferación los transcritos se expresan de manera semejante entre las muestras infectadas con los tipos virales 16 y 18, mientras que los de ciclo celular muestran un alto grado de sobrexpresión en las muestras VPH16 y una supresión en las VPH18; excepto en CaLO que muestra un patrón similar a VPH16. Esto ocurre también en el grupo de "metabolismo de fármacos", que está conformado principalmente por genes que actúan en la regulación del estado "redox" en las células y en transportadores de diferentes compuestos; en este grupo el aumento en la abundancia relativa de estos transcritos en las células VPH16 y en CaLO puede estar indicando una selección para la resistencia a fármacos.22 Este resultado puede sugerir que los genes de respuesta a fármacos son activados en presencia del VPH16 en las pacientes.
El grupo denominado de componente tumoral, está compuesto por genes tales como: homólogo de homeobox 2, proteína de tumor D52 y la H2A miembro L, que son genes que han sido frecuentemente mencionados de tener alterada su expresión en diversos tipos de tumores y que el GO les ha asignado esta categoría.
Como se esperaba, en el grupo de transcritos que el programa Gene Ontology clasificó como genes pertenecientes a procesos estromales tales como: proteína de membrana epitelial 1, trombospondina 1, Calponina 3, caldesmonina 1 y el colágeno tipo IV alfa 1, entre otros; los tejidos tumorales muestran un incremento en su expresión probablemente debido al componente de estroma presente en el tumor. Sin embargo, SiHa y CaSki presentaron el mismo incremento, esto pudiera explicarse debido a la presencia de transcritos de un gran grupo de genes de colágeno.
El grupo de "interferón" y algunos genes asociados que son regulados por éstos, indican patrones de expresión diferentes entre todas las muestras sugiriendo variación en la respuesta de la ruta de interferones. Nuestros resultados apoyan a resultados previamente publicados.23
Por otro lado, con el propósito de identificar posibles marcadores moleculares de esta neoplasia, se analizaron genes que estuvieran sobrexpresados en todas las muestras independientemente del tipo viral presente. Un gran número de esta clase de genes son los ribosomales, cuya sobrexpresión en los tumores ya ha sido previamente reportada y puede reflejar un aumento del metabolismo en células malignas, además de que esta sobrexpresión se presenta en tumores bien diferenciados. Los resultados de los experimentos de expresión mediante RT–PCRs indican y corroboran los niveles de sobrexpresión de IL6 y MMP10 en las líneas celulares y las biopsias previamente mostrados en el experimento de microhileras.
Como se esperaba por reportes previos, uno de los genes sobrexpresados fue el de la IL6; este gen codifica una citocina que, además de regular algunos aspectos de la respuesta inmune, tiene actividad inflamatoria24 y es el principal factor autocrino de crecimiento de algunas neoplasias hematológicas, especialmente aquellas derivadas de linfocitos B.25 Además, se ha sugerido tener una función de mediador central en la inflamación de infecciones del tracto genital femenino.26 Es posible que en CaCU funcione también como factor de crecimiento, lo cual concuerda con el hecho de que algunas líneas celulares de esta neoplasia expresan el receptor de IL6, aunque la relación funcional no se ha probado formalmente, además de que en cuanto a algunas de las líneas celulares utilizadas en este estudio aún no se ha examinado la expresión de IL6R.
En el cervix uterino existen muchos tipos de células capaces de elevar su nivel de expresión como son los linfocitos, macrófagos, fibroblastos, queratinocitos, células endoteliales y células tumorales.27 Además, se ha comprobado que los niveles de IL6 se incrementan en las secreciones cérvico–vaginales de las pacientes con CaCU y su producción incrementa con respecto a la severidad de la neoplasia.28 Por otro lado, existen reportes que indican que esta interleucina principalmente es producida en células tumorales y que además puede funcionar como un factor de crecimiento en líneas celulares cervicales.29 De ahí que la sobrexpresión de este gen valida los experimentos realizados. En cuanto a la sobrexpresión del gen MMP 10 encontrada en este trabajo, existen diferentes reportes que relacionan a las MMPs con algunos cánceres;30 sin embargo, actualmente no existe ningún reporte que la asocie con CaCU, lo que muestra que este gen puede ser relevante en la carcinogénesis cervical y además indica que algunos estudios deberían ser dirigidos en esta dirección.
La infección por VPH muestra patrones característicos de expresión con marcadas diferencias con respecto a los tejidos VPH negativos. El análisis mostró diferencias en la expresión de los genes IL6 y MMP 10 en los tejidos VPH(+) y VPH(–). Estos resultados podrían contribuir a conocer parcialmente (una fotografía molecular) el proceso invasor del cuello uterino, indicando, además, que las alteraciones en citocinas y componentes de matriz extracelular están estrechamente relacionadas con la carcinogénesis cervical influida por VPHs–AR.
AGRADECIMIENTOS
Al Dr. Alberto Monroy (FES–Zaragoza, UNAM) por proporcionar las líneas celulares CaLO e INBL, al Dr. D. Arenas (CMN SXXI–IMSS) por su apoyo. Este trabajo fue parcialmente apoyado por No. 34686–M y 7114 (Fondos Sectoriales, CONACyT–México). GV, PP, AH, CP fueron becarios de CONACYT, DGEP–UNAM e IMSS.
REFERENCIAS
1. World Health Statistics, 1987–1990. World Health Organization. [ Links ]
2. Programa de prevención y control del cáncer cérvico–uterino. México: Secretaría de Salud; 1992. [ Links ]
3. Brisson J, Morin C, Fortier M, et al. Risk factors for cervical intraepithelial neoplasia: differences between low– and high–grade lesions. Am J Epidemiol 1994; 140: 700–10. [ Links ]
4. Castellsague X, Bosch F, Muñoz N. Environmental co–factors in HPV carcinogenesis. Virus Research 2002; 89: 191–9. [ Links ]
5. Schiffman M, Bauer H, Hoover R, Epidemiologic evidence showing that human papillomavirus infection causes most cervical intraepithelial neoplasia. J Nati Cancer Inst 1993; 85: 958–64. [ Links ]
6. Muñoz N. Human papillomavirus and cancer: the epidemiological evidence. J Clin Virol 2000; (1–2): 1–5. [ Links ]
7. Walboomers J, Jacobs M, Manos M, Bosch F, Kummer K, et al. Human Papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189: 12–19. [ Links ]
8. Bosch F, Lorincz A, Muñoz N, Meijer C, Shah K. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol 2002; 55: 244–65. [ Links ]
9. Lorincz A, Temple G, Kurman R, Jenson A, Lancaster W. Oncogenic association of specific human papilloma virus types with cervical neoplasia. J Int Cancer Inst 1987; 79: 671–7. [ Links ]
10. Choo KB, Chen CM, Han CP, Cheng WT, Au LC. Molecular analysis of cellular loci disrupted by papillomavirus 16 integration in cervical cancer: frequent viral integration in topologically destabilized and transcriptionally active chromosomal regions. J Med Virol 1996; 49: 15–22. [ Links ]
11. Miinger K, Werness BA, Dyson N, Phelps WC, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J 1989; 8: 4099–4105. [ Links ]
12. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990; 63: 1129–36. [ Links ]
13. Park T, Fujiwara H, Wright T. molecular biology of cervical cancer and its precursors. Cancer 1995; 76(Suppl): 1902–13. [ Links ]
14. Milde–Langosch K, Riethdorf S. Role of cell–cycle regulatory proteins in gynecological cancer. J Cell Physiol 2003; 196: 224–44. [ Links ]
15. Shyu JS, Chen CJ, Chiu CC, Huang SC, Harn HJ. Correlation of human papillomavirus 16 and 18 with cervical neoplasia in histological typing and clinical stage in Taiwan: an in–situ polymerase chain reaction approach. J Surg Oncol 2001; 78: 101–9. [ Links ]
16. Caceres–Cortes J, Alvarado–Moreno J, Waga K, Rangel–Corona R, Monroy–Garcia A, et al. Implication of tyrosine kinase receptor and steel factor in cell density–dependent growth in cervical cancers and leukemias. Cancer Res 2001; 61: 6281–9. [ Links ]
17. Eisen M, Spellman P, Brown P, Botstein D. Cluster analysis and display of genome–wide expression patterns. Proc Nati Acad Sci 1998; 95: 14863–8. [ Links ]
18. Davies J, Garrod D. Molecular aspects of the epithelial phenotype. Bioessays 1997; 19: 699–704. [ Links ]
19. Ross D, Scherf U, Eisen M, Perou C, Rees C, Spellman P, Iyer V, Jeffrey S, Rijn M, Walthman M, Pergamenschikov A, Lee J, Lahkari D, Shalon D, Myers T, Weinstein J, Botstein D, Brown P. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genetics 2000; 24: 227–35. [ Links ]
20. Ahn WS, Bae SM, Lee JM, Namkoong SE, Han SJ, Cho YL, Nam GH, Seo JS, Kim CK, Kim YW. Searching for pathogenic gene functions to cervical cancer. Gynecol Oncol 2004; 93: 41–8. [ Links ]
21. Welsh J, Zarrinkar P, Sapinoso L, Kern S, Behling CA. Analysis of gene expression in normal and neoplasic ovarian tissue samples identifies candidate molecular markers of epithelial ovarian cancer. Proc Nati Acad Sci 2001; 3: 1176–81. [ Links ]
22. Moscow J, Schenider E, Ivy S, Cowan K. Multidrug resistance Chemoter Biol Response Modif 1997; 17: 139–77. [ Links ]
23. Nguyen H, Hiscott J, Pitha P. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev 1997; 8: 293–312. [ Links ]
24. Kishimoto T. The biology of Interleukin–6. Blood 1998; 74: 1–10. [ Links ]
25. Hoffman–Liebermann B, Liebermann DA. Supression of c–myc and c–myb is tightly linked to terminal differentiation induce by IL6 or LIF and not growth and no inhibition by myeloid leukemia cells. Oncogene 1991; 6: 903–9. [ Links ]
26. Rochter H, Holley R, Andrews W, Owen J, Miller K. The association of interleukin 6 with clinical and laboratory parameters of acute pelvic inflammatory disease. Am J Obstet Gynecol 1999; 181: 940–4. [ Links ]
27. Espevikn T, Wange S, Faxvaag A, Shalaby M. Regulation of interleukin–2 and interleukin–6 production from T–cells: involvement of interleukin–1 beta and transforming growth factor beta. Cell Immunol 1990; 126: 4756–61. [ Links ]
28. Tjiong M, van der Vange N, Kate F, Tjiong SP, Ter Shegget J. Increased IL–6 and IL–8 levels in cervicovaginal secretions of patients with cervical cancer. Gynecol Oncol 1999; 73: 285–91. [ Links ]
29. Eustace D, Han X, Gooding R, Riches P, Hyderman E. Interleukin–6 functions as an autocrine growth factor in cervical carcinoma in vitro. Gynecol Oncol 1993; 50: 15–9. [ Links ]
30. Gaiotto MA, Focchi J, Ribalta JL, Stavale JN. Comparative study of MMP–2 (matrix metalloproteinase 2) immune expression in normal uterine cervix, intraepithelial neoplasias, and squamous cells cervical carcinoma. Am J Obstet Gynecol 2004; 190: 1278–82. [ Links ]

