











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La tuberculosis (TB) es una enfermedad infecciosa causada por Mycobacterium tuberculosis (M. tuberculosis) y actualmente sigue constituyendo un problema de salud pública en el mundo. De acuerdo con los informes de la Organización Mundial de la Salud la cuarta parte de la población mundial está infectada y en el 2017 se registraron 10 millones de nuevos casos y 1.6 millones de muertes por esta enfermedad.1 La principal forma de la enfermedad es la pulmonar (TBP) ya que M. tuberculosis ingresa a los alvéolos pulmonares evadiendo las barreras físicas y químicas del tracto respiratorio. En los alvéolos M. tuberculosis evade los mecanismos bactericidas de los macrófagos alveolares como es la producción de péptidos antimicrobianos, las especies reactivas de oxígeno (ERO), el óxido nítrico, las citocinas proinflamatorias IL-1β, el TNF-α y la IL-12, el factor de crecimiento TGF-β y la quimiocina CXCL8.2 Los macrófagos tienen un papel crucial en los mecanismos de defensa del hospedero ya que forman parte de la inmunidad innata y participan en el desarrollo de una respuesta inmune específica para controlar la infección por M. tuberculosis y el progreso a la forma activa de la enfermedad. En la TB, la protección está asociada con la inducción de una respuesta de los linfocitos T cooperadores (Th) tipo 1 o Th1, caracterizada por producción de las citocinas proinflamatorias como IL-2, IL-12, IL-18, IFN-γ y TNF-α. La fagocitosis de M. tuberculosis por monocitos o macrófagos induce la producción de la IL-12 que favorece la producción de IFN-γ posterior a la interacción con su receptor (IL-12R) expresado por los linfocitos T.3,4 El IFN-γ es producido principalmente por las subpoblaciones de linfocitos T CD4+, CD8+ y la interacción del IFN-γ con su receptor (IFN-γR) expresado en la superficie de los macrófagos induce más de 200 genes involucrados en la activación celular, la presentación de antígenos y de los mecanismos bactericidas como la producción de óxido nítrico, la producción de ERO y de TNF-α que participan en la eliminación de patógenos intracelulares como M. tuberculosis.4,5 La importancia del IFN-γ en el control de infecciones por patógenos intracelulares se ha evidenciado en pacientes con susceptibilidad mendeliana a enfermedades causadas por micobacterias (MSMD, Mendelian Susceptibility to Mycobacterial Disease) que presentan mutaciones en el eje IL-12/IL-23/IFN-γ causando alteraciones en la producción o en la respuesta a IFN-γ.6-8 Nuestro grupo ha reportado que las células mononucleares de sangre periférica de pacientes con TBP activa producen niveles elevados de IL-10 y niveles bajos de IFN-γ en respuesta a la proteína de 30 kDa de M. tuberculosis; sin embargo, después de la terapia anti-TB la expresión del IFN-γ se incrementa a medida que disminuye la carga bacteriana por efecto del tratamiento.9 Los principales mecanismos de control de la infección por M. tuberculosis descritos en el humano incluyen la producción de péptidos antimicrobianos como las β-defensinas,10 de la catelicidina LL-37 inducible por la vitamina D,11-13 de los receptores de la inmunidad innata (TLR y NOD) y el proceso de autofagia, siendo este último un mecanismo fisiológico utilizado por las células para eliminar o reutilizar algunos componentes intracelulares, la cual también participa en la eliminación de patógenos intracelulares. En macrófagos alveolares humanos, la activación de los receptores intracelulares NOD1 y NOD2 favorece la producción de citocinas proinflamatorias, LL-37 y el aumento en la expresión de proteínas de autofagia como Atg9, Atg16L1, IRGM, LC3 y p62, con una función relevante en la eliminación de M. tuberculosis.14,15 La producción de péptidos antimicrobianos y la autofagia (IRGM) son inducibles por la activación de la inmunidad innata, pero también pueden ser inducidos por IFN-γ.16 Dada la importancia del IFN-γ se ha utilizado en aerosol aunado al tratamiento anti-TB de los pacientes con TBP17,18 pero los resultados son controversiales probablemente porque los procesos fisiológicos son complejos y existen mecanismos que aún se desconocen.
Objetivo
En el presente estudio evaluamos el efecto del IFN-γ exógeno en macrófagos alveolares humanos infectados con M. tuberculosis H37Rv en la inducción de la expresión del péptido antimicrobiano LL-37 y la proteína de autofagia IRGM (mecanismo bactericida inducido por IFN-γ), la producción de TNF-α (citocina inducida por IFN-γ) y el control del crecimiento intracelular de M. tuberculosis.
Métodos
Grupo de estudio
Se reclutaron 14 sujetos sanos de 25 ± 3 años, ocho hombres, seis mujeres, 13 de ellos PPD negativos y uno PPD positivo, 12 no fumadores y dos fumadores, sin ningún tipo de patología pulmonar o sistémica. Los voluntarios otorgaron su consentimiento por escrito para participar en el proyecto. Se determinó que eran sanos por los resultados de las pruebas de laboratorio clínico de química sanguínea completa, biometría hemática, pruebas de funcionamiento hepático, pruebas de coagulación, colesterol y triglicéridos, serología para VIH, rayos X de tórax y la revisión por un médico participante del protocolo. Este protocolo fue revisado y aprobado por los Comités de Ciencia y Ética en Investigación del Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas (INER).
Obtención de macrófagos alveolares
A los voluntarios se les realizó una broncoscopia con lavado bronquioalveolar (LBA) en el Servicio de Broncoscopia del INER. Después de anestesia local de vías aéreas superiores con lidocaína al 2% se introdujo un broncoscopio óptico flexible (P30, Olympus BF, New Hyde Park, NY) hasta el lóbulo medio y se instilaron 180 ml de solución salina estéril con recuperación inmediata. El porcentaje de recuperación del lavado fue de 74.4 ± 13.5 (media ± DE). El LBA fue centrifugado a 1,200 rpm por 15 min a 4 oC y las células bronquioloalveolares resuspendidas a una concentración de 1 x 106 células/ml en medio de cultivo RPMI-1640 (Lonza-Biowhittaker, Walkersville, MD) suplementado con 50 μg/ml de sulfato de gentamicina (Lonza-Biowhittaker), 2 mM L-glutamina (Lonza-Biowhittaker) y 10% de suero humano AB inactivado por calor (Valley Biomedical Products & Services Inc.) al que en adelante se denomina sólo medio. La suspensión celular contenía el 90.1 ± 8.6% de macrófagos, determinado por morfología y la viabilidad de las células fue determinada por exclusión de azul de tripano (Invitrogen Corporation, Grand Island, NY) siendo en general de 98%.
Cultivo de M. tuberculosis
La cepa virulenta de M. tuberculosis H37Rv (ATCC # 25618) fue cultivada en agitación durante 21 días en medio líquido 7H9 suplementado con 10% de ADC (Difco Laboratories, Detroit, MI). Se cosechó en tubos de 50 ml (Corning Costar Co., Corning, NY) y se centrifugó a 3,000 rpm durante 15 minutos a temperatura ambiente. Se eliminó el sobrenadante y el paquete bacteriano se resuspendió en medio 7H9-ADC. Se hicieron alícuotas de 1 ml y se almacenaron a -70 oC. Posteriormente, para determinar la concentración del stock se hicieron diluciones seriadas 1:10 en el medio 7H9-ADC y se sembraron en placas de agar 7H10-OADC. Después de 21 días de incubación se cuantificaron las unidades formadoras de colonias (UFC) y la micobacteria se utilizó en los ensayos de infección.
Ensayos de infección con M. tuberculosis
Las células bronquioloalveolares (0.2 x 106/pozo) se dejaron adherir durante 1 h a 37 oC y 5% de CO2 en una placa de cultivo de 96 pozos de fondo plano de poliestireno (Corning Costar Co., Corning, NY) y después de remover las células no adherentes mediante lavado con medio del cultivo, los macrófagos alveolares fueron infectados con M. tuberculosis H37Rv resuspendida en medio infectante (medio con 30% con suero humano AB inactivado por calor) a multiplicidad de infección (MOI) de 0.1, 1 y 10 y se incubaron durante 1 h. Para eliminar las micobacterias no fagocitadas las células se lavaron con medio y posteriormente se cultivaron en medio con 10% de suero humano AB inactivado por calor.
Crecimiento intracelular de M. tuberculosis
Para evaluar el control del crecimiento intracelular de M. tuberculosis H37Rv, los macrófagos infectados se incubaron durante 1 h (D0), 24 h (D1) y 96 h (D4). A los tiempos indicados las células se lisaron con SDS (dodecil sulfato de sodio) al 1% seguido por la adición de albúmina sérica bovina al 20% y con los lisados celulares se realizaron diluciones seriadas 1:10 en medio 7H9-ADC y se sembraron por triplicado en agar 7H10-OADC. Después de 21 días de incubación a 37 oC y 5% de CO2 se contaron las UFC.
Cultivos celulares de macrófagos estimulados con IFN-γ recombinante humano (rhIFN-γ)
En una placa de cultivo de 24 pozos de poliestireno (Corning Costar Co., Corning, NY) se colocaron 0.5 x 106 células por pozo y fueron estimuladas con 100 UI/ml de rhIFN-γ (R&D System Inc., McKinley Place NE, MN) o 100 ng/ml de lipopolisacárido (LPS, Sigma-Aldrich Chemical Co.) en presencia o ausencia de M. tuberculosis H37Rv a MOI 0.1, 1 y 10 y se incubaron durante 24 h a 37 oC y 5% de CO2. Se recuperaron los sobrenadantes y posteriormente fueron utilizados para determinar la producción de TNF-α por el método de ELISA. Mientras que las células adheridas en la placa se lisaron con 350 μl de la solución de lisis RLT (Qiagen Co., Strasse, Alemania) y se almacenaron a -20 oC para después hacer la extracción del RNA.
Expresión de LL-37 e IRGM por PCR en tiempo real
Se realizó la extracción de RNA total por columna a partir de los lisados celulares utilizando el RNAeasy Mini Kit (Qiagen Co.) de acuerdo con las instrucciones del fabricante. Después se realizó la síntesis del cDNA utilizando el kit Superscript-First-Strand cDNA Synthesis System (Invitrogen, Carlsbad, CA) siguiendo las instrucciones del fabricante. Brevemente, el RNA, dNTPs y los hexámeros fueron desnaturalizados (65 oC, 5 min) y se adicionó la mezcla de transcripción reversa (25 mM MgCl2, 2 mM DTT, RNAse Out inhibitor, PCR buffer 10X) y la síntesis se llevó a cabo con la enzima Superscript II en el termociclador Verity (Applied Biosystem, Foster City, CA). El cDNA se utilizó para evaluar la expresión de los genes de interés mediante la técnica de PCR (Polymerase Chain Reaction) cuantitativo en tiempo real (qPCR) utilizando los ensayos Taqman (Applied Biosystem, Foster City, CA) para los genes LL-37 (CAMP, Hs00189038 m1), IRGM (Hs01013699 s1) y el RNA ribosomal 18S como gen constitutivo. Para evaluar la expresión de los genes de interés se hizo una cuantificación relativa que utiliza el método del 2(-ΔΔCt) una vez que previamente se ha evaluado la eficiencia de la amplificación para cada ensayo. La expresión de cada gen se normalizó con el gen constitutivo y se reportaron los cambios en la expresión del gen de interés relativa a las células sin estimular.19
Cuantificación del factor de necrosis tumoral (TNF-α)
La producción del TNF-α por los macrófagos se determinó en los sobrenadantes de cultivo por la técnica de ELISA, utilizando los anticuerpos antihumano TNF-α humano purificado y antihumano. Anti-TNF-α conjugado a biotina y a continuación se utilizó la enzima estrepto-avidina-fosfatasa alcalina (Jackson ImmunoResearch, West Grove, PA) y se agregó el sustrato. La reacción se detuvo y la absorbancia fue determinada a 405 nm en un lector de placas. Las muestras se procesaron por duplicado y los resultados fueron reportados en pg/ml.20
Análisis estadístico
Las diferencias estadísticas para la expresión de los genes de LL-37 e IRGM se determinaron por una prueba de columna para muestras relacionadas de Wilcoxon y las diferencias en la producción de TNF-α y las UFC se compararon con la prueba para más de tres muestras relacionadas entre sí, la ANOVA de Friedman, seguida del postest de Dunn. Se consideró una significancia estadística cuando el valor de p < 0.05. Utilizamos el GraphPad Prism Software, Versión 6.0, 2015.
Resultados
Expresión de genes de moléculas antimicrobianas inducidas por el rhIFN-γ
Dentro de los principales mecanismos antimicrobianos de los macrófagos humanos se encuentra la producción de la catelicidina LL-37 y la autofagia (IRGM, Immunity Related GTPase M) en respuesta al rhIFN-γ. Encontramos que la adición de rhIFN-γ a macrófagos alveolares no causó cambios significativos en la expresión de LL-37, pero sí en la expresión de IRGM (mediana: 1.93 [rango 0.03-231.8]; Figuras 1A y 1B). Mientras que el LPS, incluido como el control positivo de activación de macrófagos, incrementó significativamente (p < 0.05) la expresión de LL-37 e IRGM (LL-37, mediana: 9.5 veces el control, [rango 0.01-62.4]; IRGM, mediana: 153.2 veces el control, [rango 0.01-725.0]; Figuras 1A y 1B). Para valorar el efecto de la adición del rhIFN-γ en macrófagos infectados con la cepa virulenta de M. tuberculosis H37Rv se utilizó la dosis baja de infección (MOI 0.1) con la finalidad de reducir el efecto de M. tuberculosis, cuya capacidad de evasión de la respuesta inmune es alta. La infección con M. tuberculosis indujo cambios no significativos en la expresión de LL-37, mientras que la expresión de IRGM se incrementó significativamente (p < 0.05) (IRGM, mediana: 3.53 veces el control, [rango 0.35-200.2]; Figuras 1C y 1D). La adición de rhIFN-γ a los macrófagos infectados causó un aumento significativo (p < 0.05) en la expresión de LL-37 (LL-37, mediana 1.93 veces el control, [rango 0.04-41.1]), pero no en la expresión de IRGM (Figuras 1C y 1D). Mientras que en los macrófagos infectados la adición del LPS aumentó significativamente (p < 0.05) la expresión de LL-37 e IRGM.

LL-37: catelicidina LL-37; IRGM: Immunity Related GTPase M; rhIFN-γ: interferón γ recombinante humano; LPS: lipopolisacárido; Mtb: M. tuberculosis H37Rv.
Figura 1: Expresión génica de LL-37 e IRGM inducida por rhIFN-γ. Los macrófagos alveolares humanos fueron estimulados con 100 UI de rhIFN-γ o 100 ng/ml de LPS durante 24 h. A y B: Los macrófagos fueron infectados con M. tuberculosis H37Rv a MOI 0.1 durante 1 h. Las micobacterias no fagocitadas se eliminaron y se añadieron 100 UI de rhIFN-γ o 100 ng/ml de LPS. Las células se incubaron por 24 h adicionales. C y D: Después del tiempo de incubación se eliminó el sobrenadante y las células se lisaron para extracción de RNA. La expresión de LL-37 e IRGM se midió por PCR en tiempo real (qPCR). Se muestran resultados individuales, n = 6. La línea punteada indica el nivel de expresión basal (medio) y las líneas horizontales indican la mediana.*p < 0.05 vs. medio.
Efecto de la estimulación in vitro con rhIFN-γ sobre la producción de TNF-α por los macrófagos
Debido a que el rhIFN-γ modula parte de los mecanismos de respuesta del macrófago alveolar infectado, investigamos si otros parámetros de activación del macrófago estaban modificados. Así que se midió uno de los marcadores importantes de la activación del macrófago como la producción de TNF-α. La adición de rhIFN-γ no estimuló la producción de TNF-α en macrófagos alveolares no infectados (mediana: 22.5 pg/ml [rango 0-1083]; Figura 2A). Los macrófagos infectados a MOI 0.1 mostraron bajos niveles de TNF-α (mediana: 170 pg/ml [rango 0-594]) y la adición de IFN-γ incrementó ligeramente su producción (mediana: 302 pg/ml, [rango 0-1704]), pero el cambio no fue significativo (Figura 2B). Para descartar que la dosis de infección fuera demasiado baja para medir la producción de TNF-α se incluyó un grupo de muestras a las que se les infectó con mayores cargas bacterianas (MOI 1 y 10). La producción de TNF-α aumentó en relación con la carga bacteriana a MOI 1 (mediana: 929 pg/ml [rango 373-1,345]) y MOI 10, (mediana: 3,385 pg/ml [rango 1,515-32,574]), Figuras 2C y 2D); sin embargo, los cambios no fueron estadísticamente significativos. Por otra parte, la adición del rhIFN-γ no modificó esta producción de TNF-α (MOI 1+ IFN-γ: mediana: 865 pg/ml [rango 404-6,548] y para MOI 10 + IFN-γ mediana: 1,524 pg/ml [rango 690-16,604]). El LPS incluido como control positivo indujo la producción de grandes cantidades de TNF-α en todas las condiciones, la producción en macrófagos no infectados fue de 2,075 pg/ml (rango 356-4,072), y en macrófagos infectados a MOI 0.1 fue de 13,557 pg/ml (rango 8,704-29,300); para MOI 1 fue de 21,466 pg/ml (rango 4,026-29,500) y para MOI 10 fue de 27,750 pg/ml (rango 2,546-39,824).

TNF-α: Factor de necrosis tumoral; rhIFN-γ: Interferón gamma recombinante humano; LPS: Lipopolisacárido; Mtb: M. tuberculosis; MOI: Multiplicidad de infección.
Figura 2: Producción de TNF-α por macrófagos infectados y tratados con rhIFN-γ. Los macrófagos alveolares fueron estimulados con 100 UI de rhIFN-γ o 100 ng/ml de LPS durante 24 h (a, n = 7). Los macrófagos fueron infectados con M. tuberculosis H37Rv a MOI 0.1 (b, n = 6), 1 (c, n = 4) y 10 (d, n = 4) durante 1 h. Las micobacterias no fagocitadas se eliminaron y se estimuló con 100 UI de rhIFN-γ o 100 ng/ml de LPS. Las células se incubaron por 24 h adicionales. Los sobrenadantes fueron recuperados y la producción de TNF-α se cuantificó por ELISA. Se muestran resultados individuales. Las líneas horizontales señalan la mediana.*p < 0.05 vs. medio.
La adición exógena de rhIFN-γ no contribuye a la actividad bactericida de los macrófagos humanos infectados con M. tuberculosis
El macrófago alveolar posee múltiples mecanismos antimicrobianos. Aunque la estimulación con rhIFN-γ indujo la expresión del péptido antimicrobiano LL-37, otros mecanismos podrían estar activados. Por lo tanto, evaluamos la capacidad del rhIFN-γ de inducir la muerte bacteriana directamente midiendo la carga intracelular de M. tuberculosis H37Rv, ya que el control de la infección se espera que ocurra dentro del macrófago. Observamos que el hrIFN-γ no indujo la actividad bactericida del macrófago alveolar contra M. tuberculosis H37Rv puesto que ni utilizando dosis bajas de infección se observó el control del crecimiento intracelular. Para el día 4 postinfección, para la MOI 0.1, la carga intracelular bacteriana de los macrófagos fue de 0.25 x 105 ± 0.13 x 105 UFC/ml y la de los macrófagos tratados con rhIFN-γ fue de 0.35 x 105 ± 0.19 x 105 UFC/ml; (Figura 3A); para MOI 1, la carga intracelular bacteriana de los macrófagos fue de 2.02 x 105 ± 1.2 x 105 UFC/ml y la de los macrófagos tratados con rhIFN-γ fue de 1.3 x 105 ± 0.48 x 105 UFC/ml (Figura 3B); mientras que en los macrófagos infectados con MOI 10, la carga intracelular bacteriana fue de 6 x 105 ± 2.76 x 105 UFC/ml y tratados con rhIFN-γ fue de 5.8 x 105 ± 2.43 x 105 UFC/ml (Figura 3C).
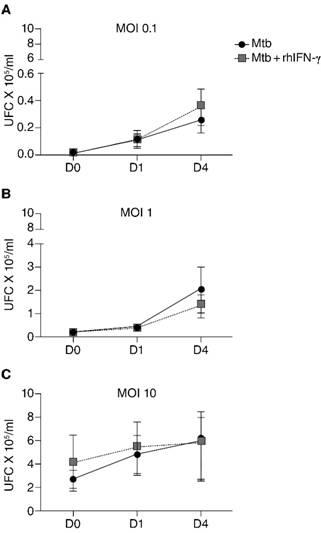
MOI: multiplicidad de infección; UFC: unidades formadoras de colonia; D0: 1 h; D1: 24 h; D4: 96 h postinfección.
Figura 3: Efecto del rhIFN-γ sobre el control del crecimiento intracelular de M. tuberculosis. Los macrófagos fueron infectados con M. tuberculosis H37Rv a MOI 0.1 (a, n = 9), 1 (b, n = 5) y 10 (c, n = 5) durante 1 h. Las micobacterias no fagocitadas se eliminaron y se añadieron 100 UI de rhIFN-γ. Las células se incubaron durante 1 h (D0), 24 h (D1) o 96 h (D4). Los sobrenadantes se eliminaron y las células se lisaron para obtener las micobacterias intracelulares. El crecimiento intracelular de M. tuberculosis H37Rv se cuantificó por triplicado por UFC. Se muestran la media ± error estándar.
Discusión
La TB es una enfermedad respiratoria de difícil control debido a que el agente causal, el bacilo M. tuberculosis, es un patógeno intracelular que evade los mecanismos inmunológicos del hospedero, entre los que se encuentran la inducción de péptidos antimicrobianos (AMP), los productos intermedios reactivos de oxígeno o nitrógeno, los productos de NADPH oxidasa y óxido nítrico sintasa (NOS2).21 Se sabe que de los mediadores solubles el IFN-γ es esencial para el control intracelular de M. tuberculosis, debido a que induce los mecanismos bactericidas intracelulares en los macrófagos de manera no redundante, así que la alteración en su producción y/o la funcionalidad de su receptor conlleva a una incapacidad para controlar la infección, lo que resulta en el desarrollo de la enfermedad activa. Lo anterior ha sido demostrado tanto en modelos de ratón deficientes de IFN-γ, así como en niños que presentan el síndrome de MSMD que afecta la funcionalidad del eje del IL-12/IL-23/IFN-γ.6-8,22 Como opción terapéutica en la deficiencia de IFN-γ, en niños con MSMD y en pacientes con TBP activa se ha sugerido la administración exógena de rhIFN-γ para reconstituir la deficiencia inmune; sin embargo, se necesitan más ensayos para evaluar su eficacia y seguridad,6-8,23 lo que sugiere que aún se desconocen los mecanismos moleculares del papel del IFN-γ en el contexto de la activación de los macrófagos alveolares humanos y el control intracelular de M. tuberculosis.
En esta investigación se evaluó la capacidad del rhIFN-γ exógeno para inducir la expresión del péptido antimicrobiano LL-37 y la proteína de autofagia IRGM. Los ensayos in vitro muestran que en macrófagos no infectados, la estimulación con de rhIFN-γ induce la expresión de IRGM, mientras que en macrófagos infectados con M. tuberculosis H37Rv, el rhIFN-γ induce la expresión de LL-37, pero no de IRGM. Debido a que el rhIFN-γ fue capaz de inducir el péptido antimicrobiano LL-37 en macrófagos infectados, se sugería que también tendría un efecto en el control intracelular de las micobacterias; sin embargo, observamos que no hay control del crecimiento intracelular a concentraciones bajas de infección (MOI 0.1) como a intermedias o altas (MOI 1 o 10). Lo anterior sugiere que el control intracelular de M. tuberculosis no depende exclusivamente de un mecanismo de inducción de LL-37 dependiente del IFN-γ tal vez se requiere la activación de la respuesta inmune adaptativa para el control de la infección por M. tuberculosis.
También es probable que IFN-γ por sí solo no sea suficiente para la activación eficiente de los macrófagos y se requiera la integración de múltiples señales, incluyendo la de los receptores tipo Toll, que no se estudiaron simultáneamente en este trabajo.24 Además, la infección con M. tuberculosis podría inhibir la actividad del péptido antimicrobiano LL-37, ya que su precursor (hCAP-18) requiere ser cortado por proteasas para ejercer su actividad antimicrobiana.25 También, es posible que el nivel de inducción de LL-37 por el IFN-γ no sea suficiente para el control del crecimiento intracelular de M. tuberculosis ya que se sabe que la expresión de LL-37 se induce hasta 100 veces por una vía dependiente de la vitamina D, cuyo uso como terapia complementaria en TB es recomendado.26
Por otro lado, después de la inducción de los mecanismos bactericidas, se requiere la activación de la respuesta inmune adaptativa, y es donde el TNF-α juega un papel importante en el control inicial y a largo plazo de la TB, así como el control de la infección durante la fase latente.27,28 En modelos in vitro se ha demostrado que la producción de TNF-α es inducida por el tratamiento con rhIFN-γ en la línea celular de macrófagos murinos RAW 264.7.29
En nuestros ensayos, la infección de macrófagos con M. tuberculosis H37Rv a MOI 0.1, 1 y 10 causó un incremento en la producción de TNF-α aunque no fue significativo, y la adición del rhIFN-γ no incrementó su producción.
Cabe mencionar que los macrófagos estimulados con otros PAMPS (Pathogen-Associated-Molecular-Patterns) como el LPS incrementaron significativamente la producción de TNF-α, lo que sugiere que esta limitación en la producción de TNF-α en los macrófagos infectados puede ser un mecanismo específico de inhibición por parte de M. tuberculosis. Aunque existe evidencia de que el TNF-α puede inducir la maduración del fagosoma-lisosoma promoviendo la actividad antimicobacteriana de manera indirecta,30 al parecer no es una vía inducida por la estimulación con el rhIFN-γ.
Estos datos demuestran que se debe considerar otros inmunoestimuladores de la inmunidad innata como la loperamida, el ácido nordihidroguayarético (NDGA) o la α-mangostina para activar los mecanismos bactericidas en los macrófagos, como hemos demostrado en nuestros trabajos previos14,15,31,32 como una alternativa al tratamiento complementario para la TB.

