











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkPresentación del caso
Presentamos el caso de una paciente del género femenino de 36 horas de vida extrauterina, originaria de la Meza de Arturo, Chihuahua. Hija de la novena gesta de madre de 36 años de edad al momento del embarazo, en apariencia sana. No llevó control prenatal adecuado, refiere ingesta de ácido fólico desde el primer trimestre de embarazo, no se realizó ningún ultrasonido durante el mismo. Cursó con embarazo de evolución normal. Nació a término vía parto eutócico, lloró y respiró al nacer, requirió sólo maniobras básicas de reanimación neonatal con un peso al nacimiento de 3,500 g, talla de 51 cm y APGAR 8-9 sin complicaciones, ingresó a alojamiento conjunto con la madre. Sin otro antecedente de importancia.
Inició su padecimiento actual durante las primeras horas de vida, posterior al inicio de la alimentación al seno materno, presentando datos clínicos de dificultad respiratoria caracterizados por aleteo nasal, tiros intercostales, disociación toracoabdominal y oximetría de pulso de 80% requiriendo iniciar manejo con oxígeno complementario con cámara cefálica a 3 L/min presentando mejoría parcial en oximetría de pulso hasta 90%. Después cursó con evolución tórpida, incrementando los datos clínicos de dificultad respiratoria, por lo que se tomó gasometría arterial, misma que reportó hipoventilación, hipoxemia grave y acidosis respiratoria, siendo necesario iniciar manejo avanzado de la vía respiratoria mediante intubación orotraqueal, se decide su traslado a la Unidad de Cuidados Intensivos Neonatales del Hospital Infantil de Especialidades del Estado de Chihuahua.
A su ingreso, la paciente llegó con apoyo respiratorio mediante cánula orotraqueal y bolsa autoinflable con presión positiva manual, clínicamente despierta, reactiva, con adecuada coloración de piel y tegumentos, normocéfalo, fontanela anterior amplia de 3.5 por 3 cm normotensa, fontanela posterior puntifome, tórax normolíneo, con presencia de tiros intercostales bajos y retracción xifoide; a la auscultación se encontraron campos pulmonares con murmullo vesicular bilateral y estertores bronquiales en hemitórax derecho, el abdomen con hepatomegalia hasta 4 cm por debajo de borde costal inferior, el resto de la exploración sin otros datos de importancia.
Se solicitó radiografía portátil de tórax en proyección anteroposterior (Figura 1) donde se observó hemitórax derecho con incremento en la distancia de los espacios intercostales con respecto a los contralaterales, con ligera herniación del parénquima pulmonar hacia la pared torácica en el séptimo, octavo y noveno espacio intercostal y hacia mediastino con desplazamiento total del mismo hacia hemitórax izquierdo; en el lóbulo superior había una imagen radiolúcida, heterogénea, hipertensa, con bordes bien delimitados que ocupa desde el primer hasta el octavo espacio intercostal derecho, con múltiples imágenes de características aparentemente quísticas en su interior.

Figura 1: Radiografía simple de tórax al ingreso. Se observa en hemitórax derecho una imagen radiolúcida, heterogénea, hipertensa, con bordes bien delimitados con desplazamiento de estructuras mediastinales a hemitórax izquierdo.
Ante los hallazgos radiológicos se estableció la sospecha diagnóstica de malformación congénita de la vía aérea pulmonar (MVAP), razón para solicitar tomografía de alta resolución simple de tórax (Figura 2) donde se observó en el parénquima pulmonar del hemitórax derecho una gran lesión de aspecto multiquístico con dimensiones de 48 × 48 × 63 mm y nivel hidroaéreo en su interior con quistes que variaban su tamaño desde 1 hasta 7 mm, hallazgos compatibles con el diagnóstico presuntivo por lo que es valorada por el Servicio de Neumología Pediátrica y Cirugía de Tórax y programada para manejo quirúrgico.
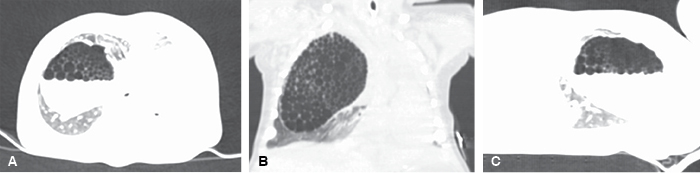
Figura 2: Tomografía de alta resolución de tórax. Se observa en hemitórax derecho una lesión grande con imágenes quísticas múltiples y nivel hidroaéreo en su interior. Panel A. Vista en proyección axial. Panel B. Vista en proyección coronal. Panel C. Vista en proyección sagital.
Se realizó toracotomía posterolateral derecha a nivel de quinto espacio intercostal, el lóbulo superior con color oscuro, con aparente pérdida de parénquima, es disecado por planos hasta localizar el bronquio para lóbulo superior, mismo que se observa y palpa con pérdida de la firmeza de la pared cartilaginosa así como con tejido bronquial circundante adelgazado. Posterior a ello, fueron localizadas las arterias y venas para el lóbulo superior, las cuales fueron ligadas y cortadas, también se cisuró de forma manual los bordes del lóbulo superior y resecado por completo el lóbulo superior derecho, colocando finalmente una sonda de drenaje pleural con sello de agua. Posterior a la recuperación posquirúrgica, el paciente reingresó a la sala de Cuidados Intensivos Neonatales para continuar manejo y tomar radiografía de tórax de control (Figura 3) donde se observó adecuada expansión de parénquima pulmonar de hemitórax derecho. Permaneció en manejo con ventilación mecánica por 72 horas, después se programó extubación electiva; sin embargo, a las 24 horas cursó con datos clínicos de dificultad respiratoria y acidosis respiratoria requiriendo manejo avanzado de la vía aérea y ventilación mecánica. Se realizó tomografía de tórax apreciando lesión quística residual (Figura 4), por lo que fue programada para nuevo evento quirúrgico y realizar resección de segmento 5 de pulmón derecho. Posterior a la cirugía reingresó a sala de Cuidados Intensivos Neonatales persistiendo inestable, continuando manejo con ventilación mecánica convencional sin presentar mejoría del estado ventilatorio y requerir manejo con ventilación de alta frecuencia oscilatoria. A las 72 horas posteriores presentó paro cardiorrespiratorio, se realizaron maniobras avanzadas de reanimación cardiopulmonar sin presentar respuesta ante las mismas.
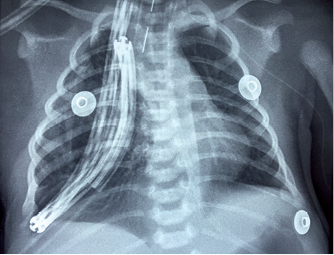

Figura 4: Tomografía de alta resolución de tórax a las 96 horas del evento quirúrgico. Se observa en hemitórax derecho una imagen quística grande, homogénea con bordes bien delimitados, neumotórax derecho con desplazamiento de las estructuras mediastinales a hemitórax izquierdo y pérdida de volumen del parénquima pulmonar derecho. Panel A. Vista en proyección axial. Panel B. Vista en proyección coronal. Panel C. Vista en proyección sagital.
La pieza quirúrgica fue enviada al área de Anatomía Patológica quienes reportaron, en la descripción macroscópica, espécimen de lóbulo superior derecho de 7 × 3 × 3 cm con superficie pleural lisa, congestiva y blanda. Al corte se identificó estructura quística mayor de 3.2 cm y una menor de 1.5 cm en su diámetro mayor, ambas, sin contenido en su interior; el resto del parénquima pulmonar congestivo, hemorrágico y de aspecto esponjoso (Figura 5).

En la descripción microscópica había quistes mayores con un revestimiento epitelial de epitelio cilíndrico pseudoestratificado, ciliado, rodeado por músculo liso y escasa isla de cartílago hialino. Alrededor de los quistes de mayor tamaño se observaron múltiples bronquiolos espalda con espalda revestidos por epitelio cilíndrico ciliado, rodeados de músculo liso sin cartílago. El diagnóstico histopatológico fue MVAP tipo II (Figura 6).
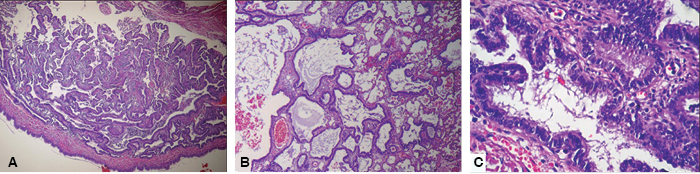
Figura 6: Imágenes microscópicas. Panel A. Imagen microscópica 4x: Se observan múltiples estructuras colapsadas con características similares a bronquiolos revestidos por un epitelio cilíndrico simple ciliado, las cuales se encuentran separadas por estructuras alveolares. Panel B. Imagen microscópica 10x: Se observan múltiples estructuras dilatadas con características similares a bronquiolos revestidos por un epitelio cilíndrico simple ciliado mismas que se encuentran separadas por estructuras alveolares. Panel C. Imagen microscópica 40x: Detalle de una estructura similar a bronquiolo en el cual se identifica el epitelio de revestimiento cilíndrico simple ciliado. La pared de dicha estructura carece de cartílago.
Discusión
La MVAP, antes llamada malformación adenomatoidea quística, es una alteración en el desarrollo de las vías respiratorias terminales, falta de desarrollo alveolar y organización inadecuada del mesénquima pulmonar. (1) Se trata de lesiones hamartomatosas que incluso pueden llegar a comprimir y repercutir en el desarrollo de los órganos vecinos. (2),(3),(4) Presenta una incidencia de 1:25,000-35,000 (2),(4),(5),(6),(7) y corresponde al 25% de las malformaciones congénitas pulmonares. La causa subyacente es desconocida. La anomalía se atribuye por lo regular a un defecto de maduración, específicamente por la detención en la fase pseudoglandular (semana 7-17 de gestación) generando una hiperplasia compensatoria con remplazo del tejido pulmonar por quistes de tamaño y distribución variable. (4),(5),(6),(7),(8)
La clasificación de Stocker describe cinco tipos: (2),(3),(6),(9)
Tipo 0: es una entidad rara representa el 1-2% de los casos. Corresponde a una displasia acinar que suele ser bilateral e incompatible con la vida.
Tipo 1: representa el 65% de los casos. Con resección quirúrgica tiene buen pronóstico. Son lesiones quísticas en su mayoría, con diámetro entre 3-10 cm rodeado de quistes de menor tamaño. A menudo son multiloculares. Los quistes tienen epitelio columnar pseudoestratificado ciliado con células productoras de mucina, la pared está compuesta por tejido elástico y fibromuscular y, a veces, por cartílago. Hay comunicación con el árbol bronquial y de manera ocasional con una arteria sistémica. El carcinoma bronquioalveolar se ha asociado con grandes malformaciones pulmonares quísticas como ésta.
Tipo 2: representa el 10-15% de los casos, por lo común se presenta en el primer año de vida. Tiene pobre pronóstico porque casi siempre va acompañado de otras anomalías congénitas, sobre todo del tracto urinario. Las lesiones se componen de quistes de tamaño medio entre 0.5-2 cm de diámetro distribuidos en tejido pulmonar normal. Por lo general afectan un solo lóbulo o parte de él, pero en ocasiones suelen afectar todo un pulmón. Hay ausencia de células productoras de mucina y cartílago, es probable que esta alteración del desarrollo sea secundaria a la obstrucción de las vías respiratorias-atresia bronquial.
Tipo 3: su aparición es infrecuente y representa sólo el 5-8% de los casos. Está compuesto por quistes menor de 0.2 cm de diámetro formando una gran masa voluminosa que abarca un lóbulo completo o incluso todo un pulmón, por lo que quizás esté asociado con desplazamiento del mediastino que los otros subtipos. Microscópicamente la lesión se asemeja a un pulmón inmaduro desprovisto de bronquios, con apariencia adenomatoidea. No se observan células mucosas, cartílago ni células rabdomiomatosas. Se asocia a atresia laríngea.
Tipo 4: representa 10-15% de los casos. Consiste en una malformación hamartomatosa del acino que consta de quistes periféricos. Pueden ser hallazgos incidentales, pero se han descrito en asociación con neumonía, neumotórax espontáneos o dificultad respiratoria. Por lo general se presenta en un solo lóbulo, rara vez es bilateral. Macroscópicamente son grandes quistes de paredes delgadas, revestidos por células aplanadas de epitelio simple aplanado o cuboidal con tejido subyacente laxo, fibrovascular y mesenquimal. (2),(3),(6),(9) Cada vez es más claro que muchas de estas lesiones son neoplásicas y representan blastomas pleuropulmonares quísticos bien diferenciados. (9),(10)
El examen clínico y la radiografía de tórax invariablemente identifican MVAP. Las manifestaciones clínicas son dificultad respiratoria progresiva que incluye: taquipnea, quejido respiratorio, retracciones y cianosis, o puede cursar asintomática. El examen físico puede mostrar abombamiento del hemitórax afectado, hiperresonancia ipsilateral, disminución de los sonidos respiratorios, ruidos cardíacos velados y contralaterales, hepatoesplenomegalia por hiperexpansión torácica. En la radiografía de tórax pueden observarse lesiones quísticas, sólidas o mixtas que afectan uno o varios lóbulos pulmonares, desviación del mediastino, derrame plural, pericárdico o neumotórax.
Mediante ultrasonografía prenatal las lesiones de MVAP se pueden identificar entre las 17-20 semanas de gestación. (2),(3),(11) La resonancia magnética también está siendo utilizada con mayor frecuencia para examinar el feto y puede ayudar a diferenciar de otras lesiones torácicas confirmando el diagnóstico en un 98% de los casos. (7),(11) La relación del volumen de la lesión (alto × ancho × longitud × 0.52) dividido por el valor de la circunferencia cefálica, mayor de 1.6 realizado mediante cualquiera de los dos métodos de imagen comentados nos ofrece un alto riesgo de desarrollar hidropesia fetal (hasta el 75-80%). (5),(8),(10),(12) La hidropesia se asocia a una mortalidad del 100% y se presenta en el 5% de los casos. (8) Dentro de los diagnósticos diferenciales se encuentran: el secuestro broncopulmonar, enfisema lobar congénito, quiste broncogénico, neumatocele, hernia diafragmática congénita, agenesia pulmonar, duplicaciones del tracto digestivo anterior del embrión y malformaciones vasculares. (4),(6),(7)
Las lesiones macroquísticas pueden producir desplazamiento del mediastino y, de manera simultánea, generar hidrops debido a la obstrucción de la vena cava y compresión cardíaca, que resulta en insuficiencia cardíaca, esto puede drenarse percutáneamente en el útero para disminuir la presión sobre las estructuras circundantes, con el inconveniente de que el líquido del quiste puede volver a acumularse, (1) además de que se ha asociado con mortalidad de 12% y obstrucción en 29% de los casos. Los riesgos adicionales a este procedimiento incluyen: ruptura prematura de membranas, hemorragia, infección, parto pre término y lesión del feto. (1) El tratamiento prenatal se considera cuando hay desplazamiento severo del mediastino, compresión del pulmón derecho, polihidramnios y ascitis en las 21 a 26 semanas de gestación. Se puede realizar shunt pleuro-amniótico o lobectomía fetal vía histeroscopia. (13) Otras técnicas son la ablación con láser o radiofrecuencia, así como escleroterapia percutánea guiada por sonografía. También se utilizan esteroides maternos cuando existe alto riesgo de hidropesia con resultados variables. (14)
La resección quirúrgica es el estándar de oro para el diagnóstico y tratamiento posnatal. La resección quirúrgica en los niños asintomáticos es más beneficiosa y con menos complicaciones en comparación con la intervención tras el desarrollo de los síntomas. (6),(13)
La lobectomía es el estándar tradicional para la resección, y es superior a la segmentectomía en lesiones unilaterales. Sin embargo, las estrategias de preservación de pulmón como la segmentectomía puede realizarse en casos de lesiones segmentarias pequeñas bien definidas y en niños con afectación bilateral o multilobar con el riesgo de presentar fugas aéreas, enfermedad residual o malignidad. (14)
La toracotomía abierta se considera el abordaje estándar para cualquier resección pulmonar; sin embargo, la aparición de técnicas mínimamente invasivas avanzadas ha llevado a la utilización cada vez frecuente de la toracoscopia(14) pues además de las ventajas cosméticas, ésta permite mejorar significativamente la discriminación entre el pulmón normal y enfermo, así como una mejor visualización de las fisuras y estructuras vasculares. Razón para apoyar la adaptación de esta técnica en personas e instituciones con conocimientos y habilidades técnicas adecuadas.
La involución y desaparición de las lesiones se han observado en 12-20% de los casos.5 El riesgo de malignidad es bajo, 0.7%.

