










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkNeumología y cirugía de tórax
versión impresa ISSN 0028-3746
Neumol. cir. torax vol.75 no.1 México ene./mar. 2016
Revisión
Primer Consenso Mexicano sobre Fibrosis Pulmonar Idiopática
First Mexican Consensus on Idiopathic Pulmonary Fibrosis
Coordinadores: Mayra Mejía,* Ivette Buendía-Roldán,‡ Heidegger Mateos-Toledo,‡ Andrea Estrada,‡ Manuel Espinoza-Hernández,§ Fortunato Juárez-Hernández,âII Claudia Vargas-Domínguez,‡ Moisés Selman¶
Colaboradores: Moisés Acuña-Kaldman, Yessica Aguilar-Navarrete, Alejandro Arreola-Morales, Alma Bizarrón-Muro, Erick Bringas-Locela, Omar Ceballos-Zúñiga, Francisco Javier Chan-Mérida, Uriel Chavarría-Martínez, Jorge Chávez-Sarabia, Alfredo Domínguez-Peregrina, Guadalupe Espitia-Hernández, Xicoténcatl García-Jiménez, Miguel Gaxiola, Alejandro Gómez-Gómez, Carlos Gómez-Zamora, Francisco González Juárez, Fernando Guillén-Ortega, Luz Audina Mendoza-Topete, Jaime Morales-Blanhir, Marcela Nájera-Cruz, Virginia Novelo-Retana, María Dolores Ochoa-Vázquez, Roberto Alfonso Perea-Sánchez, Paulo Pinaya-Ruiz, Enrique Rivero-Aguilar, Julio Robledo-Pascual, Lya Pensado-Piedra, Sergio Romero-Díaz, Alma Vázquez-García
*Jefe de Servicio de Neumología, INER;
‡Neumólogo, INER;
§Neumólogo, IMSS Cd Juárez;
IIMédico Radiólogo, INER;
¶ââââââDirector de Investigacion, INER.
Trabajo recibido: 12-I-2016; aceptado: 14-I-2016
Palabras clave: Fibrosis pulmonar idiopática, tomografía computada de alta resolución, neumonía intersticial usual, discusión multidisciplinaria.
Key words: Idiopathic pulmonary fibrosis, high-resolution computed tomography, usual interstitial pneumonia, multidisciplinary discussion.
Abreviaturas:EPI: Enfermedad pulmonar intersticial.
ETC: Enfermedad del tejido conjuntivo.
FPI: Fibrosis pulmonar idiopática.
INER: Instituto Nacional de Enfermedades Respiratorias.
OR: Odds Ratio.
ADN: Ácido desoxirribonucleico.
ATS: American Thoracic Society.
ERS: European Respiratory Society.
JRS: Japanese Respiratory Society.
ALAT: Asociación Latinoamericana del Tórax.
NIU: Neumonía intersticial usual.
TCAR: Tomografía computada de alta resolución.
BPQ: Biopsia pulmonar quirúrgica.
DMD: Discusión multidisciplinaria.
NHc: Neumonitis por hipersensibilidad crónica.
NII: Neumonía intersticial idiopática.
LBA: Lavado bronquioloalveolar.
NINE: Neumonía intersticial no específica.
CTD-ILD: Connective tissue disease-interstitial lung disease.
EA-FPI: Exacerbación aguda en fibrosis pulmonar idiopática.
HP: Hipertensión pulmonar.
HR: Hazard ratio.
PSAP: Presión sistólica de la arteria pulmonar.
mmHg: Milímetros de mercurio.
ERGE: Enfermedad por reflujo gastroesofágico.
SAHS: Síndrome de apnea-hipopnea del sueño.
NAC: N-acetilcisteina.
CVF: Capacidad vital forzada.
FDA: Federal Drug Administration.
COFEPRIS: Comisión Federal para la Protección contra Riesgos Sanitarios.
DLCO: Difusión pulmonar de monóxido de carbono.
C6M: Caminata de 6 minutos.
PDGF: Factor de crecimiento derivado de las plaquetas.
VEGF: Factor de crecimiento vascular endotelial.
FGF: Factor de crecimiento de los fibroblastos.
EPOC: Enfermedad pulmonar obstructiva crónica.
FPI: Fibrosis pulmonar idiopática.
VNI: Ventilación no invasiva.
IRA: Insuficiencia respiratoria aguda.
NT-proBNP: Propéptido natriurético cerebral.
CPAP: Presión positiva continua de la vía aérea.
CO2: Bióxido de carbono.
ENFERMEDAD PULMONAR INTERSTICIAL
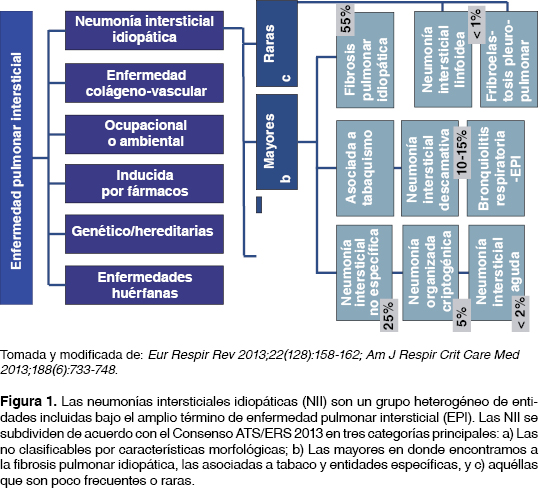
La enfermedad pulmonar intersticial (EPI) en ausencia de algún proceso infeccioso y/o neoplásico engloba varios trastornos respiratorios, los cuales tienen presentación clínica variable (aguda, subaguda o crónica). Debido a que son entidades con histologías diferentes, cursan con diferentes grados de inflamación y fibrosis que al progresar muestran rasgos cicatrizales variables donde el pulmón terminal es el resultado final de esta progresión, perdiendo sus características específicas, lo que imposibilita diferenciarlas en forma adecuada. Se reconocen alrededor de 150 entidades bajo este término; sin embargo, debido a la gran heterogeneidad de las patologías englobadas en este rubro, para su mejor entendimiento se les ha agrupado de acuerdo con características comunes que comparten entre ellas; siendo así que la clasificación tradicional se enfoca primordialmente en las asociaciones conocidas subyacentes que incluyen: a) enfermedades del tejido conjuntivo (ETC); b) enfermedades granulomatosas; c) exposiciones ambientales; d) inducidas por drogas; e) entidades relacionadas a tabaquismo; f) y por supuesto, el grupo de las neumonías intersticiales idiopáticas; en este último grupo es donde se encuentra una de las patologías más comunes que es la fibrosis pulmonar idiopática (FPI), la cual representa un poco más de la mitad (≈55%) de los casos de EPI como se muestra en la figura 1.1,2
FIBROSIS PULMONAR IDIOPÁTICA
Definición
La FPI es una forma específica de neumonía intersticial fibrosante de causa no conocida, la cual presenta en forma tradicional una evolución crónica, progresiva e irreversible; característica clínica que le confiere un pronóstico grave y letal, por lo que se le considera una enfermedad devastadora con supervivencia media reportada en las diferentes series de tan sólo 3 a 5 años después de emitirse el diagnóstico de la misma (figura 2).3-5

Epidemiología
La FPI representa una de las entidades diagnosticadas más frecuentes en los centros de referencia de EPI a nivel internacional. En México, este mismo patrón de presentación se conserva en el Instituto Nacional de Enfermedades Respiratorias (INER), la FPI es una de las tres causas más comunes de morbilidad y mortalidad de EPI. Aunque es considerada una entidad poco frecuente, en la literatura representa entre 17-86% en la práctica neumológica.6-10 En general, aunque la mayor cantidad de información epidemiológica proviene de Estados Unidos y Europa,11-14 los datos sobre incidencia, prevalencia y mortalidad en FPI son aún escasos y varían en los diferentes estudios, por lo que la incidencia y prevalencia reales de la enfermedad se desconocen, pero parece que son mayores que las reportadas a nivel mundial, debido a que se ha observado un posible aumento en su identificación en los últimos años. Sin embargo, aún son pocos los estudios enfocados a conocer más sobre su epidemiología, por lo que se estima que las tasas globales de prevalencia oscilan entre los 14 y 43 casos por 100,000 habitantes/año, aunque esta tasa tiende a incrementarse con la edad, mientras que las tasas de incidencia oscilan entre 7 y 16 por 100,000 habitantes/año.15,16 Entre los datos más confiables tenemos que en Reino Unido se reporta una incidencia anual aproximada de 8 a 9 casos por 100,000 habitantes, lo que representaría más de 5,000 casos nuevos por año.17 En México y Latinoamérica no contamos con información epidemiológica que nos permita estimar la magnitud del problema, lo más cercano a nuestra población son los datos reportados en 2013 por la normativa española para el diagnóstico y tratamiento de la FPI, donde estimaron para ese año que la FPI afecta a unas 7,500 personas en España.18 De acuerdo con los datos publicados, si tomamos en cuenta las cifras estimadas y transpolamos éstas a la población mexicana, consideramos que los posibles casos de FPI en nuestro país oscilarían alrededor de unas 10,000 personas afectadas por esta entidad.
En forma independiente a los datos reportados en los diferentes países, se ha observado que el incremento en la prevalencia, probablemente es debido a la optimización de los métodos diagnósticos y al aumento de la esperanza de vida en la población a nivel mundial. En México este número será mayor en los próximos años tomando en consideración que la esperanza de vida pasó de 60 años en el siglo pasado a 75 años en este siglo.
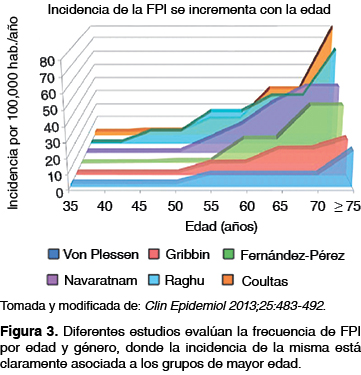
Son muchos los estudios que evalúan las tasas de FPI por edad y género, los cuales han demostrando claramente que la FPI es más frecuente en los grupos de mayor edad, hallazgo que es consistente con el papel del envejecimiento en la patogénesis de la misma.19 La FPI es una entidad que se presenta principalmente en personas de mediana edad a adultos mayores (> 55 años), siendo rara en pacientes jóvenes (por debajo de los 45 años) e inexistente en la infancia, por lo que su diagnóstico debe sospecharse principalmente en adultos entre la sexta y séptima década de vida (figura 3).1,3 En relación con el género, la FPI también parece ser más común en hombres en comparación con las mujeres,11,12 se postula que esto puede estar asociado a los patrones históricos del tabaquismo como un factor de riesgo del desarrollo de esta entidad.
Cuadro clínico
En la clínica, la FPI presenta una evolución insidiosa, crónica y progresiva, se caracteriza por la presencia de disnea y tos seca "irritativa", estos síntomas respiratorios suelen estar en forma inicial asociados al esfuerzo, pero con el tiempo pueden llegar a incapacitar al paciente, lo que se debe a la declinación de la función pulmonar en forma progresiva (tabla 1) reduciendo de esta manera la calidad de vida y conduciendo a la muerte por la presencia de insuficiencia respiratoria.

Los signos al examen físico que apoyan el diagnóstico son la presencia de estertores crepitantes en 90% de los pacientes e hipocratismo digital en 50%.20
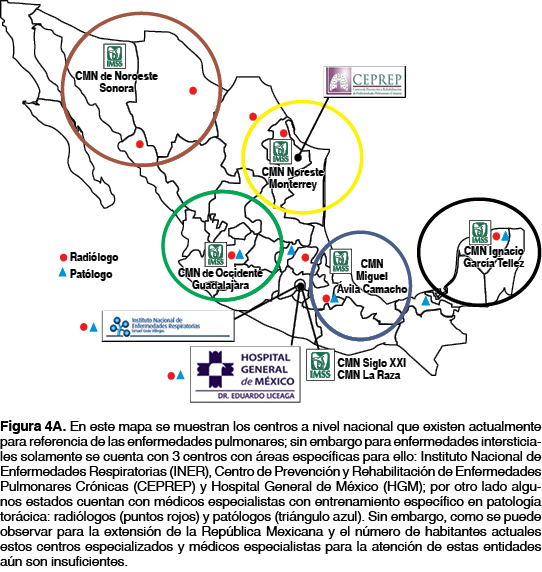
La demora entre el inicio de la sintomatología y el diagnóstico final es muy variable y puede retardarse entre 6 y 24 meses; en nuestro país se ha reportado un promedio de 18 meses para el envío de un paciente con esta patología a un centro de referencia para establecer el diagnóstico definitivo de FPI, lo que conlleva que el diagnóstico sea generalmente en etapas avanzadas de la enfermedad; una limitante que se ha identificado a través de encuestas a neumólogos es el no identificar suficientes áreas de apoyo diagnósticas como radiólogos y patólogos expertos en EPI21 (figura 4A).
Historia natural de la FPI
La supervivencia promedio en FPI es de 3 años aproximadamente a partir del momento en que se realiza el diagnóstico, se reconoce que hay un amplio espectro en la evolución clínica de la misma y ésta se asocia al pronóstico, que en la mayoría de las ocasiones es impredecible, ya que sólo cerca de 20% de los pacientes con FPI tendrán una supervivencia mayor de 5 años, esta observación resalta cómo el ritmo de progresión de la enfermedad varía entre individuos y el pronóstico individual es difícil de estimar al momento inicial del diagnóstico, ya que suele ser aparente sólo después de un período de seguimiento cuidadoso; la incertidumbre en el comportamiento de la enfermedad es desconcertante para los pacientes y sus cuidadores y pondera aún más la importancia de establecer un diagnóstico confiable antes de impartir las implicaciones que ello conlleva.
Esto se debe a que la FPI es una entidad heterogénea que se asocia a diferentes formas en su patrón de progresión,22 siendo el más común el de los progresores lentos, quienes presentan en forma característica un cuadro clínico sintomático de varios meses a años con períodos de relativa estabilidad, los que pueden asociarse a episodios de declinación acelerada (exacerbación aguda de la FPI) y que con frecuencia resultan en gran discapacidad funcional e incluso, en una alta proporción de los casos en muerte por la presencia de insuficiencia respiratoria grave.4,19 Lo que es una realidad, es que de acuerdo con el comportamiento clínico el paciente con FPI puede presentar varios escenarios de progresión y se desconoce si las diferentes formas en la historia natural representan fenotipos diferentes de una misma entidad (figura 5).19,23,24

Factores de riesgo
Aunque la FPI es una enfermedad de causa no conocida, se han identificado varios factores de riesgo asociados a la misma como: género masculino, el cual casi duplica la posibilidad de presentarla (OR1.7 [IC95%1.03-2.7]); de igual manera el riesgo casi se triplica con la exposición ocupacional a humos, químicos o gases (OR 2.8 [IC95%1.5-5.5]), tabaquismo (OR2.7[IC95%,1.7-4.4]) y reflujo gastroesofágico (OR 3.1[IC95% 1.7-5.9]);23,25 mientras que el riesgo más elevado pero menos reportado es la presencia de antecedente familiar de fibrosis pulmonar (FP), lo que lo incrementa nueve veces (OR 9.1 [IC95% 3.7-22.4]).26 Esta entidad también se asocia al envejecimiento como un factor relacionado con diferentes acontecimientos que suceden durante esta etapa de la vida como: la inestabilidad genómica, el acortamiento de telómeros ampliamente estudiado en FPI en los últimos años, disfunción mitocondrial, senescencia celular, agotamiento de células madre, comunicación intercelular alterada y diferentes alteraciones epigenéticas.27 Incluso en el último año se ha sugerido que la FPI se debe a un envejecimiento pulmonar acelerado explicado por el acortamiento de telómeros relacionado con la lesión de células epiteliales alveolares en pacientes con una arquitectura genética susceptible para desarrollar la enfermedad como son: mutaciones en TERT (5p15), MUC5B (11p15), TERC (3q26), OBFC1 (10q24) entre otras; cambios en la metilación del ADN (hipermetilación o hipometilación); senescencia inmune con incremento de células T CD4+/CD28- y autoanticuerpos circulantes; alteración en la regulación de micro-RNAs así como sobrerregulación de vías de señalización de Wnt.28
Diagnóstico
En México se debe realizar el diagnóstico de FPI siguiendo los criterios del consenso internacional del ATS/ERS/JRS/ALAT del 2011-2015:3,29 a) exclusión de otras causas conocidas de EPI, b) la presencia de un patrón de neumonía intersticial usual (NIU) en tomografía computada de alta resolución de tórax (TCAR) sin la necesidad de corroboración histológica y c) la combinación específica del patrón tomográfico y morfológico en biopsia pulmonar quirúrgica (BPQ).
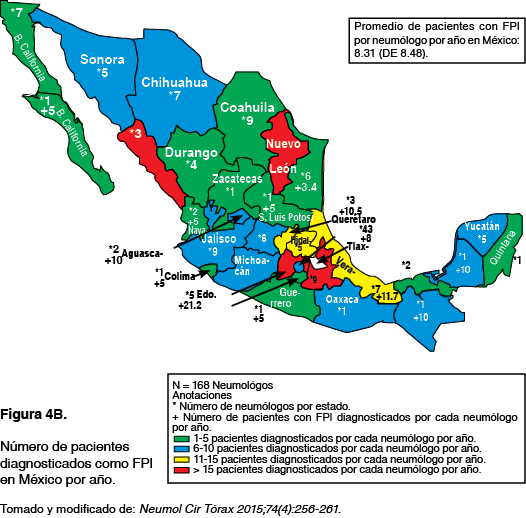
La FPI es una enfermedad de difícil diagnóstico y con frecuencia requiere la colaboración de un grupo de médicos expertos en EPI; (neumólogo, radiólogo y patólogo) para alcanzar un diagnóstico en consenso a través de una discusión multidisciplinaria (DMD), la cual incrementa su certeza diagnóstica; de esta manera el diagnóstico de la FPI representa un reto en general para el clínico quien debe identificar las redes de apoyo y centros de referencia cercanos a su práctica diaria (figura 4B).21
Es importante enfatizar que la FPI es una enfermedad que se limita a los pulmones y se asocia con un patrón de NIU en la imagen radiológica e histológica.3 A partir de 1998 Katzenstein et al.30 propusieron que el término de FPI se reserve sólo para aquellos pacientes con este patrón y sin una causa identificable de otra EPI, lo que fue aceptado y unificó el nombre para esta entidad a partir del consenso ATS/ERS de 2000.23 Sin embargo, es importante tener en cuenta que el patrón NIU no es exclusivo de FPI y puede presentarse en diferentes escenarios clínicos, por ejemplo: la ETC, toxicidad por drogas, neumonitis por hipersensibilidad crónica (NHc), entre otras causas de neumonía intersticial fibrótica (tabla 2).

Patrón tomográfico
La imagen de TCAR permite realizar un diagnóstico de FPI con la suficiente certeza sin necesidad de corroboración histológica, los datos publicados apoyan la especificidad de la misma para identificar en una alta proporción un patrón histológico de NIU (> 90%),31,32 por lo que este método de imagen se ha convertido en un componente esencial para el diagnóstico de la misma; además permite limitar los posibles diagnósticos diferenciales de acuerdo con imágenes, patrones y distribución de las alteraciones a nivel intersticial.
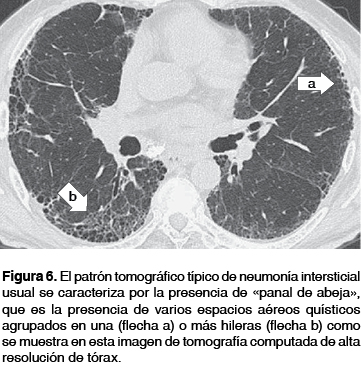
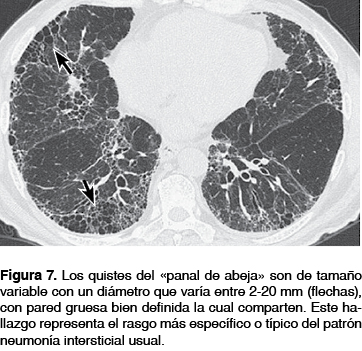
En la gran mayoría de los casos de FPI para realizar un diagnóstico definitivo es necesario encontrar la presencia de una imagen en "panal de abeja", que es la presencia de varios espacios aéreos quísticos agrupados en una o más hileras (figura 6), quistes de tamaño variable con un diámetro entre 2-20 mm, con pared gruesa bien definida y la cual comparten (figura 7), este hallazgo representa el rasgo más específico o típico del patrón NIU y su presencia se reporta de 24 a 91% de los casos con FPI; la distribución del mismo es hacia las regiones basales y posteriores de los pulmones, presentando un gradiente de menor a mayor profusión en forma apico-basal y anteroposterior, lo que da una imagen en propela, imagen que tiene un valor predictivo positivo del patrón histológico de NIU de 90 a 100%; (figura 8).

Para realizar el diagnóstico de NIU en TCAR se deben de cumplir los siguientes criterios de acuerdo con el Consenso 2011-20153,29 ATS/ERS/JRS/ALAT: a) lesiones de predominio basal y subpleural; b) opacidades de tipo reticular; c) imagen de "panal de abeja", con o sin bronquiectasias por tracción y d) ausencia de características incompatible con NIU, así estos hallazgos tomográficos se consideran al día de hoy suficientes para establecer un diagnóstico de FPI sólo cuando se han descartado otras causas de EPI.
La ausencia de "panal de abeja", pero con la presencia de opacidad reticular basal, subpleural y bilateral (figura 9) permite realizar el diagnóstico de posible patrón NIU, en donde la corroboración histólogica es necesaria para poder emitir un diagnóstico definitivo de FPI.33 Sin embargo, debido a la heterogeneidad de la enfermedad no siempre es sencillo esta evaluación, por lo que una vez que se considera que estamos ante la presencia de una neumonía intersticial idiopática (NII), los resultados de las imágenes de TCAR y los hallazgos en el estudio de histopatología se vuelven cruciales para establecer un diagnóstico lo suficientemente seguro a través de una DMD.

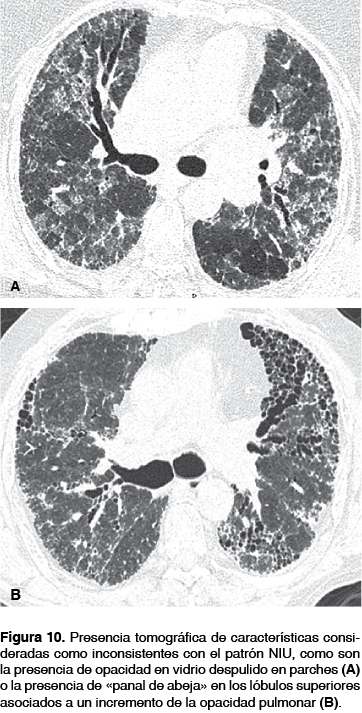
La presencia tomográfica de características consideradas como inconsistentes con el patrón NIU como son: lesiones de predominio en las partes superiores o medias del pulmón, predominio peribroncovascular, opacidades en vidrio despulido, las cuales tienen a ser más extensas que las opacidades reticulares, micronódulos profusos, quistes pulmonares (no "panal de abeja"), atrapamiento aéreo difuso y consolidación lobar o segmentaria deben alertar al radiólogo torácico sobre diagnósticos diferenciales, ya que la presencia de una o más de estas características resulta en un patrón radiográfico inconsistente con NIU (figura 10).3,34,35
El Consenso 2011-2015 reconoce la importancia en la certeza diagnóstica por imagen al identificar un patrón de NIU en un contexto clínico compatible, donde se puede obviar realizar BPQ; pero enfatiza la realización de la misma en cualquier caso que por imagen de TCAR se presente un patrón posible o inconsistente sin una causa identificable.3,29
Patrón histopatológico
La especificidad de un patrón NIU en TCAR es similar al patrón histológico de NIU, siendo poco probable la presencia de otra alteración a nivel pulmonar, por lo que la recomendación en estos casos es que la BPQ puede omitirse de acuerdo con el criterio clínico.2,3 Cuando la imagen de TCAR muestra un patrón de posible o inconsistente con patrón NIU, siempre es recomendable realizar BPQ, sobre todo cuando es necesario un diagnóstico definitivo de FPI.
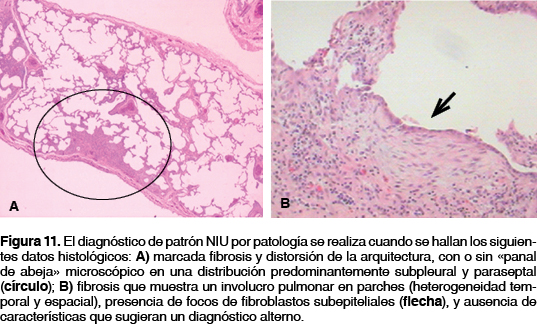
Se emite un diagnóstico de patrón NIU por patología cuando se hallan los siguientes datos histológicos: a) marcada fibrosis y distorsión de la arquitectura, con o sin "panal de abeja" microscópico en una distribución predominantemente subpleural y paraseptal; b) fibrosis que muestra un involucro pulmonar en parches (heterogeneidad temporal y espacial); c) presencia de focos de fibroblastos subepiteliales; y d) ausencia de características que sugieran un diagnóstico alterno (figura 11).
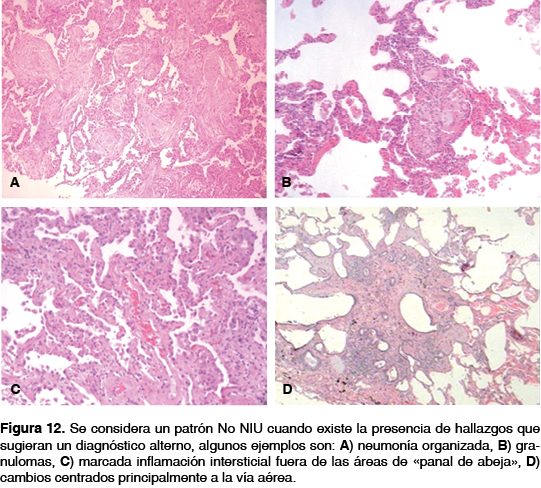
El patrón No-NIU se describe en morfología cuando los hallazgos que se consideran inconsistentes se observan en la misma, como son la presencia de: a) membranas hialinas; b) neumonía organizada; c) granulomas; d) marcada inflamación intersticial fuera de las áreas de "panal de abeja"; e) cambios centrados principalmente a la vía aérea, y f) otros rasgos que sugieran un diagnóstico alterno (figura 12).
Mientras que el patrón de probable NIU está definido por marcada fibrosis y distorsión de la arquitectura pulmonar con o sin "panal de abeja", en ausencia de otro involucro en parches o focos de fibroblastos, en ausencia de características inconsistentes con patrón NIU.
El patrón de probable NIU puede ser definido, además, como cambios de "panal de abeja" sólo detectado a través de la BPQ, reflejando un muestreo de áreas con fibrosis en estado terminal. En cambio, un patrón histológico de posible NIU requiere fibrosis difusa y en parches con o sin inflamación intersticial, en ausencia de otras características requeridas para definir un patrón de NIU definitiva y en ausencia de rasgos que sugieran un diagnóstico alterno.
Cuando se presenta un patrón de fibrosis en una EPI, el cual no encaja con ninguna de las categorías antes mencionadas se le ha denominado fibrosis no clasificable.2,3
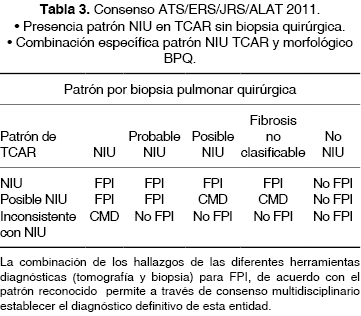
Cuando se realiza la DMD, es necesario para establecer el diagnóstico de FPI la presencia radiológica y/o histológica de un patrón definitivo de NIU; sin embargo, en algunas ocasiones no se presenta el patrón clásico de NIU ni en imagen de TCAR y/o patología, siendo importante evaluar ambos estudios para definir el diagnóstico final en cada paciente,4 como se muestra en la tabla 3.
La morfología de la NIU secundaria puede ser idéntica a la morfología de la FPI, en este caso la distinción se basa principalmente en las características clínicas y de laboratorio. Aunque existen hallazgos en el contexto de un patrón de NIU que pueden sugerir una ETC o NHc subyacente, estas alteraciones pueden ser sutiles y se deben buscar en forma intencional por el patólogo (tablas 4 y 5).36


Lavado bronquioloalveolar
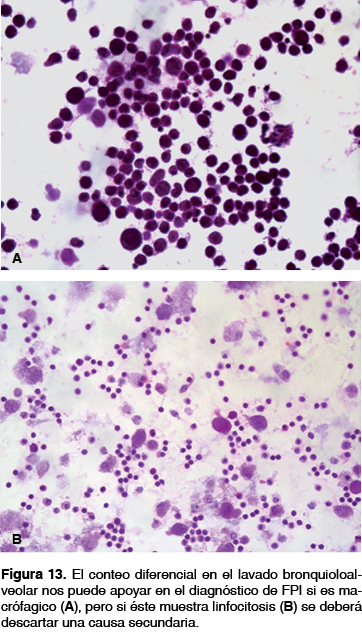
Aunque en muchos centros de referencia para EPI no se recomienda el uso del lavado bronquioloalveolar (LBA) en forma rutinaria para el diagnóstico de FPI, en México este método se utiliza con relativa frecuencia debido a su gran utilidad en las formas fibróticas de enfermedades inflamatorias que cursan con un patrón similar a NIU en TCAR y/o BPQ, en especial por la alta frecuencia de formas crónicas de neumonía intersticial no específica (NINE) y neumonitis por hipersensibilidad (NH), que son los diagnósticos diferenciales obligados antes de emitir el de FPI. Tanto la NINE como la NHc cursan con un recuento celular alveolar de predominio linfocítico (> 30%); por ejemplo, en NHc se ha reportado que al usar el LBA la reclasificación de estos casos es de 8 a 42% de las veces, aun cuando inicialmente fueron asignados a tener FPI basados en los criterios del consenso ATS/ERS/JRS/ALAT 2011 (figura 13).37-39
Biopsia transbronquial
La biopsia transbronquial no ha mostrado utilidad en el diagnóstico de FPI, debido a que la distribución de las lesiones en las áreas afectadas están en la periferia pulmonar y en parches, además, la temporalidad de las lesiones tiende a ser heterogénea, haciendo a este método diagnóstico no útil para la toma de tejido pulmonar para su evaluación morfológica, incluyendo además que su tamaño dificulta una evaluación adecuada, por lo que la toma de biopsia transbronquial se reserva para enfermedades de distribución central o linfático en donde el rendimiento diagnóstico es mayor.17 Por otro lado, la incorporación de la criobiopsia podría ser de mayor utilidad, pero en México se necesita contar con centros donde se pueda realizar y contar con la suficiente experiencia para su aplicación diagnóstica que determinen su posible utilidad en FPI.40
Diagnóstico diferencial
La FPI, al ser una entidad del grupo de las NII, su diagnóstico diferencial cobra importancia, ya que requiere estrictamente la presencia de un patrón de NUI en TCAR y/o BPQ, así como la ausencia de características que apoyen una causa secundaria. Por lo que los clínicos deben estar alertados a sospechar otras entidades aun cuando los datos clínicos no sean concluyentes, como recién ha sido reportado por el ERS/ATS Task Force on Undifferentiated Forms of CTD-ILD 41 antes de concluir un diagnóstico de FPI; aun cuando la TCAR y la BPQ muestran un patrón de NIU, ya que un porcentaje variable de estos casos pueden estar asociados a ETC.42 Hoy, el diagnóstico diferencial de la FPI demanda una responsabilidad de la comunidad neumológica en México, ya que tiene implicaciones de tratamiento y pronóstico.
Exacerbaciones
La exacerbación aguda en fibrosis pulmonar idiopática (EA-FPI) se define como el deterioro rápido, clínicamente significativo de causa no identificable que transforma la progresión insidiosa de la enfermedad en un daño agudo, se caracteriza comúnmente por un daño pulmonar agudo/síndrome de insuficiencia respiratoria aguda que puede provocar la muerte del paciente43 (figura 4). La incidencia reportada a 1 año es de 14% y a 3 años de 21%;44 siendo más frecuente en temporada de invierno y primavera, probablemente debido a infecciones virales, además, se han descrito disparadores de estos eventos como son: el lavado bronquioloalveolar, BPQ o criobiopsia, microaspiraciones (reflujo gastroesofágico), índice de masa corporal alto y niveles altos de KL-6.45,46
Los signos que se incrementan durante la exacerbación son: disnea, taquipnea y cianosis; sin embargo son inespecíficos, por lo que se han establecido criterios diagnósticos como se muestra en la tabla 6 para definir a la EA-FPI. En general, las EA-FPI tienen un pobre pronóstico a corto plazo con mortalidad de > 50% y ésta incrementa a 90-100% si se requiere apoyo mecánico ventilatorio. Entre los factores de peor pronóstico asociados están: altos niveles séricos de KL-6, baja oxigenación, mayor extensión en cambios tomográficos y enfermedad difusa en TCAR (figura 14).

El tratamiento debe incluir medidas de soporte (oxígeno suplementario, rehabilitación pulmonar, etc.), corticoesteroides a dosis altas, tratamiento de causas reversibles identificadas.47
Comorbilidades
Las comorbilidades son entidades que pueden provocar un deterioro clínico y con ello modificar el pronóstico de la enfermedad, siendo importante por ello realizar un interrogatorio exhaustivo de los antecedentes del paciente con FPI para encontrar las posibles causas que puedan generar una caída de la función pulmonar de una forma más acelerada. A continuación se describen las comorbilidades con mayor impacto en FPI.
Enfisema pulmonar y FPI
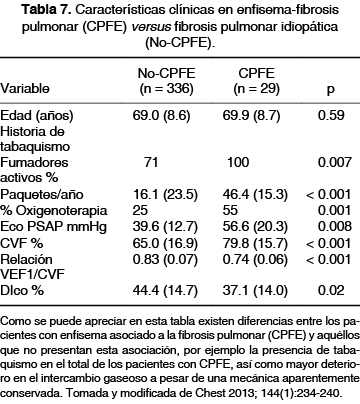
El tabaquismo es el principal factor de riesgo para desarrollar enfisema pulmonar; se ha observado que un índice tabáquico mayor de 30 paquetes por año confiere un mayor riesgo de presentar la combinación de enfisema y FPI, considerada poco frecuente; pero cada vez se detecta más desde el uso de la TCAR como herramienta diagnóstica y se reconoce que hasta 50% de los pacientes con FPI presentan evidencia radiológica de enfisema,48 aunque sólo 10% de ellos tendrán el involucro parenquimatoso suficiente para ser clínicamente significativa (> 10% del volumen pulmonar) esta asociación.49 Las alteraciones pulmonares que se encuentran son los datos ya descritos de FPI más la presencia de enfisema centrolobulillar y/o paraseptal.50 Al realizar las pruebas de función pulmonar, en el síndrome combinado el patrón mixto u obstructivo son los datos espirométricos más comunes, no así la restricción pulmonar como en la EPI clásica, estas alteraciones mecánicas deben corroborarse con la pletismografía corporal. Sin embargo, en esta asociación la mecánica pulmonar puede estar pseudonormal por la combinación de enfermedades, mientras que las pruebas de intercambio gaseoso sí logran mostrar el daño estructural que presenta el paciente, por lo que la evaluación real debe realizarse con la caminata de seis minutos y la difusión de gases (tabla 7).49
Los pacientes con enfisema y FPI presentan una mayor mortalidad debido a la alta frecuencia con que desarrollan hipertensión pulmonar (HP) y gravedad de la misma en etapas más tempranas de su evolución, en la población mexicana también se presenta este comportamiento como se muestra en la figura 15.51,52
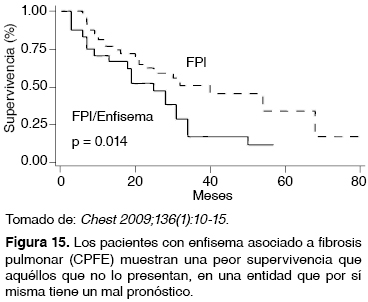
Hipertensión pulmonar
La prevalencia de HP complicando el curso de pacientes con FPI se ha reportado entre 32-85%.53 La presencia de HP se ha asociado a un riesgo mayor de mortalidad (HR para muerte, HP vs. no HP de 4.03, IC 95% 1.17-27.9).51 Asimismo, en México, en un estudio en el INER se observó que la presencia de HP grave (≥ 75 mmHg) se asocia significativamente a mortalidad (HR 2.25, IC 95% 1.12-4.54).52 De acuerdo con la clasificación de Dana Point, 2008,54 el tipo de HP pertenece al grupo 3 (HP secundaria a enfermedad pulmonar y/o hipoxemia) y ésta es determinante pronóstico. La condición patológica se considera que está dada por: 1) vasoconstricción arterial pulmonar asociada a hipoxemia crónica; 2) daño estructural con lecho vascular disminuido; y 3) progresión de las lesiones vasculares pulmonares debido a citoquinas inflamatorias y otros mediadores.55
En pacientes con disnea desproporcionada es conveniente realizar un electrocardiograma y un ecocardiograma transtorácico en búsqueda de HP. En caso de encontrar una forma grave de HP (PSAP > 46 mmHg) se debe descartar tromboembolia pulmonar asociada. El único tratamiento que ha sido estudiado en dos ensayos clínicos en pacientes con HP asociada a FPI es utilizando sildenafil (inhibidores de fosfodiesterasa-5 (PDE-5)) en donde ambos mostraron mejoría en la capacidad de ejercicio (calidad de evidencia baja).56,57 Otros fármacos como el bosentán, epoprostenol no cuentan con evidencia científica que apoye su uso en FPI.58,59
Hasta el momento no existen datos concretos que apoyen un tratamiento farmacológico. En caso de documentar hipoxemia en reposo o en la caminata se sugiere iniciar tratamiento con oxígeno en domicilio de forma crónica de acuerdo con la titulación que requiera cada paciente.
Cáncer pulmonar
La FPI comparte múltiples vías moleculares con el cáncer (epigenética, apoptosis retardada, comunicación celular disminuida, expresiones anormales mRNA), si bien no se considera una enfermedad neoplásica, se ha propuesto que se trata de una enfermedad neoproliferativa del pulmón. Al igual que para el desarrollo de FPI, uno de los principales factores de riesgo de cáncer es el tabaquismo, en donde a mayor índice tabáquico mayor riesgo de desarrollar cáncer (>20 paquetes año); al compartir el mismo factor de riesgo, los pacientes exfumadores con un índice tabáquico > 30 paquetes año tienen una incidencia siete veces mayor de cáncer pulmonar comparada con población general, lo cual le confiere un peor pronóstico.60,61 Se sugiere que en todo paciente con FPI que presente un deterioro clínico como parte de los diagnósticos diferenciales debe considerarse esta entidad, en caso de encontrar aumento de vidrio despulido y nódulos periféricos debe realizarse el abordaje diagnóstico para descartar cáncer pulmonar.62
Enfermedad por reflujo gastroesofágico (ERGE)
La ERGE es más común en FPI que en la población general,63 se estima que más de 90% de los pacientes con FPI tienen ERGE,64 la presencia de microaspiraciones nocturnas puede provocar una exacerbación de la enfermedad al desarrollar neumonitis química.65 Los cambios anatómicos que provoca la FP pueden desencadenar una dilatación esofágica empeorando la ERGE y con ello la FP.66,67 Se sugiere que a todo paciente con FPI se indique tratamiento para enfermedad por reflujo (inhibidores de la bomba de protones, procinéticos, etc.).68,69
Síndrome de apnea-hipopnea del sueño (SAHS)-obesidad
La evaluación del incremento en el índice de masa corporal (sobrepeso-obesidad) de los pacientes con FPI es un problema de salud creciente en México;70 lo cual se asocia a un aumento en la incidencia de SAHS en este grupo de pacientes que aumenta la posibilidad de hipoxemia durante el dormir, incrementando el riesgo de padecer enfermedades cardiovasculares. Por ello se deben realizar cuestionarios y escalas clínicas (EPWORTH, Berlín, SACS, etc.) para documentar la probabilidad de riesgo de SAHS en pacientes con FPI71 y obesidad e iniciar un tratamiento oportuno para mejorar la calidad de vida de los pacientes. El tratamiento ideal es el uso de ventilación no invasiva domiciliaria.
Ansiedad-depresión
La presencia de una enfermedad crónica, progresiva y mortal es causa de eventos de ansiedad generalizada y depresión en pacientes con FPI, estas comorbilidades son causa de que el paciente suspenda su tratamiento de la enfermedad de base así como de sus comorbilidades.72 Es conveniente que si a un paciente se le diagnostica FPI sea valorado por el área de psicología clínica, para identificar la presencia de ansiedad o depresión e iniciar un tratamiento psicológico oportuno. En México no existe la cultura de acudir con un psicólogo clínico; sin embargo, en enfermedades respiratorias ya se ha descrito que el tratamiento psicológico es útil para mejorar el apego al tratamiento.
Tratamiento actual de la FPI
Durante varias décadas se han probado diferentes tratamientos para la FPI con resultados no satisfactorios, el tratamiento más ampliamente utilizado a base de corticoesteroides y citotóxicos como la azatioprina durante muchos años73 mostró resultados negativos en el estudio PANTHER que comprendió tres ramas comparando la triple terapia clásica (prednisona, azatioprina, n-acetilcisteina -NAC-) y la NAC como monoterapia, contra placebo. En 2011 un análisis preliminar mostró que la triple terapia fue más deletérea para la evolución de los pacientes incrementando la mortalidad e ingresos hospitalarios que los pacientes que recibieron placebo, por lo que se suspendió su uso y en la actualidad este tratamiento no se recomienda en pacientes con FPI. La continuación del estudio con NAC vs. placebo no demostró un beneficio en la preservación de la capacidad vital forzada (CVF) en pacientes con enfermedad leve-moderada, por lo que al presente su uso como monoterapia no es recomendado.74
Desde 2014 la FDA y en el transcurso de 2014-2015 la COFEPRIS en México aprobaron el uso de dos medicamentos considerados antifibróticos solamente para FPI que son: pirfenidona y nintedanib.
La pirfenidona es una piridona con efectos antiinflamatorios y antioxidantes, antagonista de los efectos del factor de crecimiento transformante β1 (TGF β1) in vitro. Se ha sugerido que actúa también como un antifibrótico alterando la expresión, síntesis y acumulación de colágeno, inhibiendo el reclutamiento, proliferación y expresión de las células productoras de matriz extracelular. En la Unión Europea los consensos realizados en 2012 y 2013 decidieron recomendar a la pirfenidona como el estándar de tratamiento de FPI en pacientes con CVF > 50% y difusión de monóxido de carbono (DLCO) > 35%; sin embargo, no fue sino hasta 2014 que la FDA decide aprobar la droga para su uso en Estados Unidos de acuerdo con los resultados del estudio ASCEND75 que se realizó en 555 pacientes con diagnóstico de FPI con CVF entre 50 y 90% y DLCO entre 30 y 90%, aleatorizados 1:1, tratados durante 52 semanas en 127 centros de diversas partes del mundo. El objetivo primario en este estudio fue observar la caída de CVF al final del tratamiento y los objetivos secundarios: supervivencia libre de progresión de enfermedad, distancia recorrida en C6M, todas las causas de mortalidad y las relacionadas al tratamiento. Los resultados de este estudio fueron que los pacientes tratados con pirfenidona redujeron significativamente la progresión de la enfermedad medida por cambios en la caída del porcentaje de CVF desde la basal, 16.5% en el grupo tratado vs. 31.8% en el grupo placebo; asimismo, se observó estabilidad en 22.7% de los pacientes que recibían pirfenidona vs. 9.7% en el grupo placebo. Además, se demostraron cambios positivos y significativos en la C6M y en el tiempo libre de enfermedad. La suspensión del tratamiento debido a efectos adversos fue de 14.4% para pirfenidona y 10.8% para placebo. Por lo que se concluyó que el perfil riesgo-beneficio es favorable en los pacientes con FPI tratados con pirfenidona.
El nintedanib es una molécula del grupo de las antitirosincinasas no selectivo, ya que inhibe tres cinasas del receptor: factor de crecimiento derivado de las plaquetas (PDGF), factor de crecimiento vascular endotelial (VEGF) y del factor de crecimiento de los fibroblastos (FGF), lo que de acuerdo con la patogénesis conocida de la enfermedad reduce el proceso fibrótico. Los resultados de los estudios INPULSIS-1 y 2 realizados en paralelo contra placebo, multinacionales, aleatorizados 3:2, con un total de 513 y 548 pacientes con FPI, respectivamente, con CVF ≥ 50% y DLCO entre 30% a 79%, con dosis de 150 mg 2 veces al día por 52 semanas; tenían como objetivo primario: evaluar la caída anual (mL/año) en CVF y como objetivos secundarios: el cambio en el cuestionario de St. George y el tiempo a la primera exacerbación aguda. Los resultados mostraron que se cumplió el objetivo primario, ya que la disminución en la caída de la CVF fue significativamente menor en el grupo que recibió nintedanib vs. el grupo placebo.
En INPULSIS-2 hubo de manera significativa menor deterioro en el cuestionario de calidad de vida de St. George y en riesgo de exacerbación aguda. Los efectos adversos reportados con mayor frecuencia en los grupos que recibieron nintedanib fueron: diarrea 61.5% vs. 18.6% en placebo (INPULSIS-1) y 63.2% vs. 18.3% (INPULSIS-2). Con estos resultados se concluyó que el nintedanib a 150 mg cada 12 horas enlentece la progresión de la enfermedad al reducir significativamente la caída de CVF a 52 semanas y se asocia con un perfil aceptable de efectos adversos.76 De acuerdo con estos datos publicados en 2014, la aprobación de nintedanib para su uso en FPI hasta la fecha se ha obtenido en varios países incluyendo México. Asimismo, se han realizado diferentes análisis de subgrupos de poblaciones de interés que continúan en tratamiento en donde estos resultados siguen favoreciendo el uso del medicamento.
Sin embargo, existen aún dudas en relación con estos medicamentos p. ej.: no se ha definido por cuánto tiempo se sostiene la respuesta favorable de estos medicamentos, no se ha realizado una comparación de uno contra el otro, así como si el uso de ambos medicamentos en forma simultánea podría ser una opción adecuada, tampoco se ha definido si se puede iniciar con cualquiera de ellos y, posteriormente, de no tener el resultado esperado se deba hacer el cambio de uno por otro, por lo que siguen las investigaciones para responder a estas interrogantes.
Además del tratamiento farmacológico es de gran importancia no olvidar tratar la hipoxemia individualizando la cantidad de oxígeno suplementario, evitar el tabaquismo, la rehabilitación pulmonar y el tratamiento de comorbilidades.
Debido a que no hay cura para la FPI y las opciones de tratamiento aún son limitadas, el trasplante pulmonar continúa siendo la mejor opción en esta enfermedad. En una revisión sistemática de Kistler se trasplantaron de 1990 a 2011 8,528 pacientes con FPI, la sobrevida promedio fue de 4.5 años, la sobrevida a un año posterior al trasplante fue de 75% y disminuyó a 59% a los 3 años, 47% a los 5 años y 24% a los 10 años.77 Si bien, en nuestro país los intentos para tener un programa de trasplante continuo no han logrado superar las limitantes como el número de donadores así como el número de centros especializados.
Rehabilitación pulmonar
Basándose en los resultados positivos que tiene la rehabilitación pulmonar en EPOC, existe un creciente entusiasmo por aplicar este tratamiento en pacientes con FPI, ya que a medida que la fibrosis avanza se presentan síntomas como: la disnea, fatiga, tos, ansiedad y depresión que deterioran la capacidad del desempeño funcional y disminuye la calidad de vida que se asocian a desacondicionamiento musculoesquelético; razón para considerar la rehabilitación pulmonar como una intervención integral con terapias diseñadas al entrenamiento muscular, la educación y los cambios en los hábitos de vida, con el fin de mejorar la condición física y psicológica del paciente y promover la adherencia a conductas para mejorar la salud a largo plazo.78-81 Aunque hoy no existen estudios que evalúen de manera objetiva el beneficio de la rehabilitación pulmonar en pacientes con FPI, por lo que hace falta evidencia científica para conocer su impacto real.
Ventilación
La ventilación no invasiva (VNI) es una medida de soporte para la insuficiencia respiratoria crónica en varias enfermedades pulmonares. Diferentes estudios han mostrado la utilidad de la VNI en algunos casos y la necesidad de ventilación invasiva (VI) en otros.82-86 Los resultados de estos estudios se resumen de la siguiente manera:
• La evolución de los pacientes con FPI en etapas avanzadas de la enfermedad que desarrollan insuficiencia respiratoria aguda (IRA) y requieren VNI o VI es muy pobre. Aunque en ocasiones el uso de la VNI se ha asociado a beneficios clínicos, principalmente en reducir la necesidad de intubación, la tasa de complicaciones y muerte en pacientes bien seleccionados.• En pacientes con FPI complicados con IRA los niveles elevados de plasma de NT-proBNP al ingreso pueden ser considerados un predictor de pobre desenlace a pesar del uso de VNI.87
• Dado el mal pronóstico de los pacientes con IRA y FPI que son intubados, el uso de VNI se debe considerar una opción compasiva para el alivio de la disnea, la elección de este método debe ser evaluada de acuerdo con cada caso considerando la opinión del paciente y familiares.
• Una opción alterna de soporte para la insuficiencia respiratoria crónica no exacerbada en pacientes con FPI fue planteada en un estudio piloto, donde se evaluaron los efectos sobre la mecánica pulmonar del flujo nasal alto (nHF rango de 16-40 L/min) debido a su mejor tolerancia comparado con CPAP, en este estudio se reportó un incremento en la amplitud de la presión y en la presión media, un descenso en la frecuencia respiratoria y el volumen minuto además de una reducción del CO2 capilar, estos efectos sugieren relacionarse con un alivio en el trabajo respiratorio. Esta opción de soporte para la insuficiencia respiratoria crónica podría ser de utilidad en la etapa pretrasplante pulmonar, o bien compasiva en la etapa terminal, pero son necesarios estudios prospectivos.88
Métodos de evaluación pronóstica
El curso clínico de la FPI es variable, lo que impacta en el tiempo de supervivencia, que en general tiende a ser mala (3-5 años).3 Se han evaluado como posibles predictores pronósticos de evolución característicos como: datos clínicos, radiológicos, funcionales, morfológicos e incluso biomarcadores,4 los cuales se han asociado a un riesgo mayor de mortalidad.
Tradicionalmente, se usan términos como "leve", "moderado", "grave", "temprano" y "avanzado" para evaluar la enfermedad, basados en pruebas de función pulmonar,89 algunos de estos parámetros como la CVF y DLco se asocian a pronóstico y son variables de desenlace utilizadas en los estudios multicéntricos, cuando la CVF está en 50-55% del predicho y una DLco de 35-40% del predicho, se considera como enfermedad leve a moderada, separándolos de los graves que deben ser considerados más para trasplante pulmonar.90
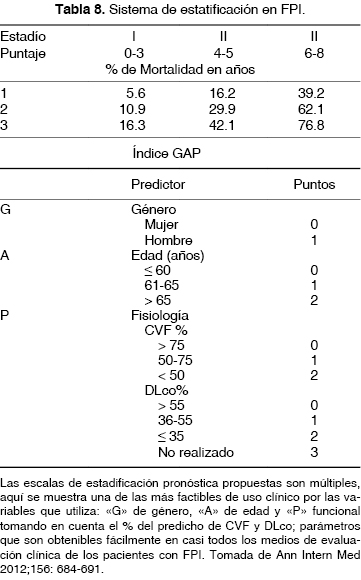
Las escalas de evaluación propuestas han sido múltiples; no obstante, muchas de ellas complejas en su uso clínico cotidiano, recién la escala de GAP91 (por sus siglas en inglés "G" de género, "A" de edad y "P" de pruebas de función pulmonar [CVF y DLco]) permite establecer tres estadíos de gravedad que se han asociado a la probabilidad de mortalidad a 1, 2 o 3 años (tabla 8). En México, actualmente se están realizando estudios con las cohortes de pacientes con FPI para validar ésta y otras escalas propuestas en la literatura.
Las direcciones futuras parecen estar más encaminadas a encontrar biomarcadores séricos, como se ha logrado en otras entidades, que nos permitan mejorar el diagnóstico, seguimiento y pronóstico de esta entidad.
CONCLUSIONES
La FPI representa el prototipo de las enfermedades fibrosantes, de evolución crónica y progresiva. La cual se observa en pacientes adultos mayores (mayores de 50 años), está limitada a los pulmones y es de causa desconocida.
Los pacientes con FPI cursan con disnea y deterioro funcional progresivo, por lo que retardar el diagnóstico les confiere un mal pronóstico (≈ 3 años, después del diagnóstico).
En los últimos años se han publicado grandes avances en la comprensión de la patogénesis, genética e historia natural de la FPI y por lo tanto en posibilidades terapéuticas específicas.92
Los resultados de los ensayos clínicos (INPULSIS y ASCEND) dan una nueva esperanza para el tratamiento de la FPI. Aunque no mostraron curar la enfermedad, sí ofrecen una reducción significativa en la progresión de la enfermedad. Ambos medicamentos están en México para el tratamiento de la FPI.
En la práctica clínica, la gestión experta del paciente con EPI también debe incluir la atención sintomática, el asesoramiento de especialistas en ejercicio y rehabilitación, evaluación y tratamiento de las comorbilidades y la derivación oportuna para ingresar a lista de trasplante.93
La TCAR ha mejorado el diagnóstico de la FPI, obviando la necesidad de la BPQ en algunos casos, sin embargo, sigue siendo de gran utilidad en un subgrupo de pacientes. Cuando no es posible realizarla por el riesgo que confiere, es necesario evaluar nuevas técnicas como la criobiopsia transbronquial, en México se están iniciando estudios y posiblemente en breve pueda evaluarse su utilidad real en pacientes con FPI.94
Los avances recientes de investigación han identificado biomarcadores moleculares como: MUC5B, KL6 y MMP7, que eventualmente pueden tener utilidad clínica y se espera puedan incorporarse como herramientas clínicas en los próximos años.95,96
El seguimiento de los pacientes con FPI debe realizarse al menos cada 6 meses para determinar la respuesta al tratamiento, ajuste de oxígeno, incremento en ejercicio y control de comorbilidades utilizando herramientas objetivas de seguimiento.
Agradecimientos
Al Laboratorio Boheringer-Ingelheim por su apoyo en las reuniones de grupo de trabajo para la realización del primer Consenso Mexicano sobre FPI.
REFERENCIAS
1. Wells AU. Managing diagnostic procedures in idiopathic pulmonary fibrosis. Eur Respir Rev 2013;22(128):158-162. http://dx.doi.org/10.1183/09059180.00001213 [ Links ]
2. Travis WD, Costabel U, Hansell DM, et al.; ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188(6):733-748. http://dx.doi.org/10.1164/rccm.201308-1483ST [ Links ]
3. Raghu G, Collard HR, Egan JJ, et al.; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183(6):788-824. http://dx.doi.org/10.1164/rccm.2009-040GL [ Links ]
4. Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183(4):431-440. http://dx.doi.org/10.1164/rccm.201006-0894CI [ Links ]
5. Bjoraker JA, Ryu JH, Edwin MK, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998;157(1):199-203. [ Links ]
6. Coultas DB, Hughes MP. Accuracy of mortality data for interstitial lung diseases in New Mexico, USA. Thorax 1996;51(7):717-720. [ Links ]
7. Karakatsani A, Papakosta D, Rapti A, et al.; Hellenic Interstitial Lung Diseases Group. Epidemiology of interstitial lung diseases in Greece. Respir Med 2009;103(8):1122-1129. http://dx.doi.org/10.1016/j.rmed.2009.03.001 [ Links ]
8. Thomeer MJ, Costabe U, Rizzato G, Poletti V, Demedts M. Comparison of registries of interstitial lung diseases in three European countries. Eur Respir J Suppl 2001;32:114s-118s. [ Links ]
9. Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med 1994;150(4):967-972. [ Links ]
10. Curbelo P. Encuesta de EPID en Latinoamérica-ALAT 2013. Respirar 2013;5: 5-8. [ Links ]
11. Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014;2(7):566-572. http://dx.doi.org/10.1016/S2213-2600(14)70101-8 [ Links ]
12. Fernández Pérez ER, Daniels CE, Schroeder DR, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest 2010;137(1):129-137. http://dx.doi.org/10.1378/chest.09-1002 [ Links ]
13. Von Plessen C, Grinde O, Gulsvik A. Incidence and prevalence of cryptogenic fibrosing alveolitis in a Norwegian community. Respir Med 2003;97(4):428-435. [ Links ]
14. Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006;61(11):980-985. [ Links ]
15. Caminati A, Madotto F, Cesana G, Conti S, Harari S. Epidemiological studies in idiopathic pulmonary fibrosis: pitfalls in methodologies and data interpretation. Eur Respir Rev 2015; 24(137):436-444. http://dx.doi.org/10.1183/16000617.0040-2015 [ Links ]
16. Ley B, Collard HR. Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol 2013;5:483-492. http://dx.doi.org/10.2147/CLEP.S54815 [ Links ]
17. Navaratnam V, Fleming KM, West J, et al. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011;66(6):462-467. http://dx.doi.org/10.1136/thx.2010.148031 [ Links ]
18. Xaubet A, Ancochea J, Bollo E, et al.; Sociedad Española de Neumología y Cirugía Torácica (SEPAR) Research Group on Diffuse Pulmonary Diseases. Guidelines for the diagnosis and treatment of idiopathic pulmonary fibrosis. Sociedad Española de Neumología y Cirugía Torácica (SEPAR) Research Group on Diffuse Pulmonary Diseases. Arch Bronconeumol 2013;49(8):343-353. http://dx.doi.org/10.1016/j.arbres.2013.03.011 [ Links ]
19. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949-1961. http://dx.doi.org/10.1016/S0140-6736(11)60052-4 [ Links ]
20. Xaubet A, Ancochea J, Blanquer R, et al.; Grupo de Investigación en Enfermedades Pulmonares Intersticiales Difusas. Área de Técnicas y Transplante. SEPAR. Diagnóstico y tratamiento de las enfermedades pulmonares intersticiales difusas. Arch Bronconeumol 2003; 39(12):580-600. [ Links ]
21. Barreto-Rodríguez JO, Mejía M, Buendía-Roldán I. Panorama actual de la fibrosis pulmonar idiopática en México. Neumol Cir Torax 2015;74(4):256-261. [ Links ]
22. Selman M, Carrillo G, Estrada A, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLos ONE 2007; 2(5):e482 [ Links ]
23. American Thoracic Society Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med 2000;161(2 Pt 1):646-664. [ Links ]
24. Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183(4):431-440. http://dx.doi.org/10.1164/rccm.201006-0894CI [ Links ]
25. García-Sancho Figueroa MC, Carrillo G, Pérez-Padilla R, et al. Risk factors for idiopathic pulmonary fibrosis in a Mexican population. A case-control study. Respir Med 2010;104(2):305-309. http://dx.doi.org/10.1016/j.rmed.2009.08.013 [ Links ]
26. García-Sancho MC, Buendía-Roldán I, Fernández-Plata MR, et al. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir Med 2011;105(12):1902-1907. http://dx.doi.org/10.1016/j.rmed.2011.08.022 [ Links ]
27. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153(6):1194-1217. http://dx.doi.org/10.1016/j.cell.2013.05.039 [ Links ]
28. Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am J Respir Crit Care Med 2014;189(10):1161-1172. http://dx.doi.org/10.1164/rccm.201312-2221PP [ Links ]
29. Raghu G, Rochwerg B, Zhang Y, et al.; American Thoracic Society; European Respiratory society; Japanese Respiratory Society; Latin American Thoracic Association. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 2015;192(2):e3-e19 http://dx.doi.org/10.1164/rccm.201506-1063ST [ Links ]
30. Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998;157(4 Pt 1):1301-1315. [ Links ]
31. Sverzellati N. Highlights of HRCT imaging in IPF. Respir Res 2013;14 Suppl 1:S3. http://dx.doi.org/10.1186/1465-9921-14-S1-S3 [ Links ]
32. Lynch DA, Godwin JD, Safrin S, et al.; Idiopathic Pulmonary Fibrosis Study Group. High-resolution computed tomography in idiopathic pulmonary fibrosis: diagnosis and prognosis. Am J Respir Crit Care Med 2005;172(4):488-493. [ Links ]
33. Sundaram B, Gross BH, Martinez FJ, Oh E, et al. Accuracy of high-resolution CT in the diagnosis of diffuse lung disease: effect of predominance and distribution of findings. AJR Am J Roentgenol 2008;191(4):1032-1039. http://dx.doi.org/10.2214/AJR.07.3177 [ Links ]
34. Wells AU. The revised ATS/ERS/JRS/ALAT diagnostic criteria for idiopathic pulmonary fibrosis (IPF)--practical implications. Respir Res 2013;14 Suppl 1:S2. http://dx.doi.org/10.1186/1465-9921-14-S1-S2 [ Links ]
35. Hodnett PA, Naidich DP. Fibrosing interstitial lung disease. A practical high-resolution computed tomography-based approach to diagnosis and management and a review of the literature. Am J Respir Crit Care Med 2013;188(2):141-149. http://dx.doi.org/10.1164/rccm.201208-1544CI [ Links ]
36. Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev 2014;23(133):308-319. http://dx.doi.org/10.1183/09059180.00004914 [ Links ]
37. Meyer KC, Raghu G, Baughman RP, et al.; American Thoracic Society Committee on BAL in Interstitial Lung Disease. An Official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med 2012;185(9):1004-1014. http://dx.doi.org/10.1164/rccm.201202-0320ST [ Links ]
38. Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009;179(11):1043-1047. http://dx.doi.org/10.1164/rccm.200808-1313OC [ Links ]
39. Morell F, Villar A, Montero MA, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med 2013;1(9):685-694. http://dx.doi.org/10.1016/S2213-2600(13)70191-7 [ Links ]
40. Babiak A, Hetzel J, Krishna G, et al. Transbronchial cryobiopsy: a new tool for lung biopsies. Respiration 2009;78(2):203-208. http://dx.doi.org/10.1159/000203987 [ Links ]
41. Fischer A, Antoniou KM, Brown KK, et al.; ERS/ATS Task Force on Undifferentiated Forms of CTD-ILD. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015;46(4):976-987. http://dx.doi.org/10.1183/13993003.00150-2015 [ Links ]
42. Moua T, Zamora Martínez AC, Baqir M, Vassallo R, Limper AH, Ryu JH. Predictors of diagnosis and survival in idiopathic pulmonary fibrosis and connective tissue disease-related usual interstitial pneumonia. Respir Res 2014;15:154. http://dx.doi.org/10.1186/s12931-014-0154-6 [ Links ]
43. Papanikolaou I, Drakopanagiotakis F, Polychronopoulos VS. Acute exacerbations of interstitial lung diseases. Curr Opin Pulm Med 2010;16(5): 480-486. http://dx.doi.org/10.1097/MCP.0b013e32833ae49d [ Links ]
44. Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356-363. http://dx.doi.org/10.1183/09031936.00159709 [ Links ]
45. Collard HR, Moore BB, Flaherty KR, et al.; Idiopathic Pulmonary Fibrosis Clinical Research Network Investigators. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2007;176(7):636-643. [ Links ]
46. Judge EP, Fabre A, Admali HI, Egan JJ. Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Eur Respir J 2012; 40(1):93-100 http://dx.doi.org/10.1183/09031936.00115511 [ Links ]
47. Antoniou KA, Wells AU. Acute exacerbation of idiopathic pulmonary fibrosis. Respiration 2013;86(4):265-274. http://dx.doi.org/10.1159/000355485 [ Links ]
48. Schmidt SL, Nambiar AM, Tayob N, et al. Pulmonary function measures predict mortality differently in IPF versus combined pulmonary fibrosis and emphysema. Eur Respir J 2011;38(1):176-183. http://dx.doi.org/10.1183/09031936.00114010 [ Links ]
49. Ryerson CJ, Hartman T, Elicker BM, et al. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest 2013;144(1):234-240. http://dx.doi.org/10.1378/chest.12-2403 [ Links ]
50. Kitaguchi Y, Fujimoto K, Hanaoka M, Kawakami S, Honda T, Kubo K. Clinical characteristics of combined pulmonary fibrosis and emphysema. Respirology 2010;15(2):265-271. http://dx.doi.org/10.1111/j.1440-1843.2009.01676.x [ Links ]
51. Cottin V, Nunes H, Brillet PY, et al; Groupe d'Étude et de Recherche sur les Maladies Orphelines Pulmonaires (GERM O P). Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J 2005;26(4): 586-593. [ Links ]
52. Mejía M, Carrillo G, Rojas-Serrano J, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest 2009;136(1):10-15. http://dx.doi.org/10.1378/chest.08-2306 [ Links ]
53. Nathan SD, Noble PW, Tuder RM. Idiopathic pulmonary fibrosis and pulmonary hypertension: connecting the dots. Am J Respir Crit Care Med 2007;175(9):875-880. [ Links ]
54. Proceedings of the 4th World Symposium on Pulmonary Hypertension, February 2008, Dana Point, California, USA. J Am Coll Cardiol 2009;54(1 Suppl):S1-S117. [ Links ]
55. Seeger W, Adir Y, Barberà JA, et al. Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol 2013;62(25 Suppl):D109-D116. http://dx.doi.org/10.1016/j.jacc.2013.10.036 [ Links ]
56. Han MK, Bach DS, Hagan PG, et al.; IPFnet Investigators. Sildenafil preserves exercise capacity in patients with idiopathic pulmonary fibrosis and right-sided ventricular dysfunction. Chest 2013;143(6):1699-1708. http://dx.doi.org/10.1378/chest.12-1594 [ Links ]
57. Zimmermann GS, von Wulffen W, Huppmann P, et al. Haemodynamic changes in pulmonary hypertension in patients with interstitial lung disease treated with PDE-5 inhibitors. Respirology 2014;19(5):700-706. http://dx.doi.org/10.1111/resp.12294 [ Links ]
58. Onda N, Tanaka Y, Hino M, Gemma A. Bosentan for pulmonary hypertension secondary to idiopathic pulmonary fibrosis. Respir Med Case Rep 2014;14:19-23. http://dx.doi.org/10.1016/j.rmcr.2014.11.008 [ Links ]
59. Hallowell RW, Reed RM, Fraig M, Horton MR, Girgis RE. Severe pulmonary hypertension in idiopathic nonspecific interstitial pneumonia. Pulm Circ 2012; 2(1):101-106. http://dx.doi.org/10.4103/2045-8932.94842 [ Links ]
60. Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med 2000; 16181):5-8. [ Links ]
61. Tomassetti S, Gurioli C, Ryu JH, et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015;147(1):157-164 http://dx.doi.org/10.1378/chest.14-0359 [ Links ]
62. National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365(5):395-409. http://dx.doi.org/10.1056/NEJMoa1102873 [ Links ]
63. Savarino E, Carbone R, Marabotto E, et al. Gastro-oesophageal reflux and gastric aspiration in idiopathic pulmonary fibrosis patients. Eur Respir J 2013; 42(5):1322-1331. http://dx.doi.org/10.1183/09031936.00101212 [ Links ]
64. Tobin RW, Pope CE 2nd, Pellegrini CA, Emond MJ, Sillery J, Raghu G. Increased prevalence of gastroesophagealnreflux disease in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998;158(6):1804-1808. [ Links ]
65. Lee JS, Song JW, Wolters PJ, et al. Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J 2012;39(2):352-358. http://dx.doi.org/10.1183/09031936.00050911 [ Links ]
66. Noth I, Zangan SM, Soares RV, et al. Prevalence of hiatal hernia by blinded multidetector CT in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2012; 39: 344-5. http://dx.doi.org/10.1183/09031936.00099910 [ Links ]
67. Tcherakian C, Cottin V, Brillet PY, et al. Progression of idiopathic pulmonary fibrosis: lessons from asymmetrical disease. Thorax 2011;66(3):226-231. http://dx.doi.org/10.1136/thx.2010.137190 [ Links ]
68. Lee JS, Ryu JH, Elicker BM, et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;184(12):1390-1394. http://dx.doi.org/10.1164/rccm.201101-0138OC [ Links ]
69. Lee JS, Collard HR, Anstrom KJ, et al.; IPFnet Investigators. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Respir Med 2013;1(5): 369-376. http://dx.doi.org/10.1016/S2213-2600(13)70105-X [ Links ]
70. Buendía-Roldan I, Mejía M, Mateos-Toledo H, et al. Valoración de peso corporal de pacientes con enfermedades intersticiales. XXVII Congreso Venezolano de Neumología y Cirugía Torácica. Porlamar, Venezuela, del 8 al 10 de julio de 2014. [ Links ]
71. Mermigkis C, Bouloukaki I, Antoniou K, et al. Obstructive sleep apnea should be treated in patients with idiopathic pulmonary fibrosis. Sleep Breath 2015; 19(1):385-391. http://dx.doi.org/10.1007/s11325-014-1033-6 [ Links ]
72. Holland AE, Fiore JF Jr, Bell EC, et al. Dyspnoea and comorbidity contribute to anxiety and depression in interstitial lung disease. Respirology 2014;19(8): 1215-1221. http://dx.doi.org/10.1111/resp.12360 [ Links ]
73. Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathriopine and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012;366(21):1968-1977. http://dx.doi.org/10.1056/NEJMoa1113354 [ Links ]
74. Idiopathic Pulmonary Fibrosis Clinical Research Network; Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2014;370(22): 2093-2101. http://dx.doi.org/10.1056/NEJMoa1401739 [ Links ]
75. King TE, Bradford WZ, Castro-Bernadini S, et al.; ASCEND study group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083-2092. http://dx.doi.org/10.1056/NEJMoa1402582 [ Links ]
76. Richeldi L, Du Bois R, Raghu G, et al.; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071-2082. http://dx.doi.org/10.1056/NEJMoa1402584 [ Links ]
77. Kistler KD, Nalysnyk L, Rotella P, Esser D. Lung transplantation in idiopathic pulmonary fibrosis: a systemic review of the literature. BMC Pulm Med 2014; 14:139. http://dx.doi.org/10.1186/1471-2466-14-139 [ Links ]
78. Nici L, ZuWallack R. Pulmonary rehabilitation: today and tomorrow. Breathe 2010;6(4):305-311. http://dx.doi.org/10.1183/18106838.0604.305 [ Links ]
79. Ries, AL, Bauldoff, GS, Carlin, BW, et al. Pulmonary Rehabilitation: Joint ACCP/AACVPR Evidence-Based Clinical Practice Guidelines. Chest 2007; 131(5 Suppl):4S-42S. [ Links ]
80. Nishiyama O, Taniguchi H, Kondoh Y, et al. Dyspnoea at 6-min walk test in idiopathic pulmonary fibrosis: comparison with COPD. Respir Med 2007; 101(4):833-838. [ Links ]
81. SwigrisJJ, Kuschner WG, Jacobs SS, Wilson SR, Gould MK. Health related quality of life in patients with idiopathic pulmonary fibrosis: a systematic review. Thorax 2005;60(7):588-594. [ Links ]
82. Blivet S, Philit F, Sab JM, et al. Outcome of patients with idiopathic pulmonary fibrosis admitted to the ICU for respiratory failure. Chest 2001;120(1):209-212. [ Links ]
83. Fumeaux T, Rothmeier C, Jolliet P. Outcome of mechanical ventilation for acute respiratory failure in patients with pulmonary fibrosis. Intensive Care Med 2001;27(12):1868-1874. [ Links ]
84. Saydain G, Islam A, Afessa B, Ryu JH, Scott JP, Peters SG. Outcome of patients with idiopathic pulmonary fibrosis admitted to the intensive care unit. Am J Respir Crit Care Med 2002;166(6):839-842. [ Links ]
85. Al-Hameed FM, Sharma S. Outcome of patients admitted to the intensive care unit for acute exacerbation of idiopathic pulmonary fibrosis. Can Respir J 2004; 11(2):117-122. [ Links ]
86. Mollica C, Paone G, Conti V, et al. Mechanical ventilation in patient with end-stage idiopathic pulmonary fibrosis. Respiration 2010;79(3):209-215. http://dx.doi.org/10.1159/000225932 [ Links ]
87. Vianello A, Arcaro G, Battistella L, et al. Noninvasive ventilation in the event of acute respiratory failure in patients with idiopathic pulmonary fibrosis. J Crit Care 2014;29(4):562-567. http://dx.doi.org/10.1016/j.jcrc.2014.03.019 [ Links ]
88. Braünlich J, Beyer D, Mai D, Hammerschmidt S, Seyfarth HJ, Wirtz H. Effects of nasal high flow on ventilation in volunteers, COPD and idiopathic pulmonary fibrosis patients. Respiration 2013;85(4):319-325. http://dx.doi.org/10.1159/000342027 [ Links ]
89. Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003;16885):538-542. [ Links ]
90. Kolb M, Collard HR. Staging of idiopathic pulmonary fibrosis: past, present and future. Eur Respir Rev 2014;23(132):220-224. http://dx.doi.org/10.1183/09059180.00002114 [ Links ]
91. Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012;156(10):684-691. http://dx.doi.org/10.7326/0003-4819-156-10-201205150-00004 [ Links ]
92. Collard HR, Bradford WZ, Cottin V, et al. A new era in idiopathic pulmonary fibrosis: considerations for future clinical trials. Eur Respir J 2015;46(1):243-249. http://dx.doi.org/10.1183/09031936.00200614 [ Links ]
93. Corte TJ, Collard H, Wells AU. Idiopathic interstitial pneumonias in 2015: A new era. Respirology 2015;20(5):697-698. http://dx.doi.org/10.1111/resp.12559 [ Links ]
94. Casoni GL, Tomassetti S, Cavazza A, et al. Transbronchial lung cryobiopsy in the diagnosis of fibrotic interstitial lung diseases. PLoS One 2014;9(2):e86716. http://dx.doi.org/10.1371/journal.pone.0086716 [ Links ]
95. Zhang Y, Kaminski N. Biomarkers in idiopathic pulmonary fibrosis. Curr Opin Pulm Med 2012;18(5):441-446. http://dx.doi.org/10.1097/MCP.0b013e328356d03c [ Links ]
96. Loomis-King H, Flaherty KR, Moore BB. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr Opin Pharmacol 2013;13(3):377-385. http://dx.doi.org/10.1016/j.coph.2013.03.015 [ Links ]
Correspondencia:
Dra. Ivette Buendía-Roldán
Servicio de Enfermedades Intersticiales del Pulmón, Instituto Nacional Enfermedades Respiratorias Ismael Cosío Villegas. Calzada de Tlalpan Núm. 4502,
Col. Sección XVI, 14080, Del. Tlalpan,
Ciudad de México, México, Tel.: 5487 1771.
Correo electrónico: ivettebu@yahoo.com.mx
Notas
Los autores declaran no tener conflicto de intereses.
Este artículo puede ser consultado en versión completa en: http://www.medigraphic.com/neumologia

