











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La enfermedad de Castleman, también conocida como hiperplasia linfoide angiofolicular, presenta variantes histológicas hialino vascular y de células plasmáticas, lo que agrega a su complejidad diagnóstica debido a su amplio espectro de presentación. Esta variedad de manifestaciones clínicas y morfológicas complica la diferenciación entre condiciones benignas, como la linfadenopatía reactiva, y cuadros malignos, como el linfoma folicular o el linfoma de la zona del manto1. Por lo tanto, el presente artículo tiene como objetivo abordar exhaustivamente el enfoque histopatológico e inmunohistoquímico de la enfermedad de Castleman y ofrecer un análisis diferencial detallado en comparación con otras entidades clínicas.
El propósito de este artículo es proporcionar una visión integral de la enfermedad de Castleman y sus desafíos diagnósticos. A través de la presentación de un caso clínico, resaltando aspectos clave del diagnóstico histopatológico e inmunohistoquímico, así como abordar el diagnóstico diferencial con otras entidades clínicas relevantes. Al realizar un análisis exhaustivo de este caso y llevar a cabo discusiones detalladas sobre los datos recopilados, el artículo contribuye a una comprensión más profunda de esta enfermedad rara y sus complejidades diagnósticas.
Al adentrarse en las complejidades de la enfermedad de Castleman, el artículo arroja luz sobre sus diversas manifestaciones clínicas y morfológicas, subrayando las dificultades para distinguirla tanto de las condiciones benignas como de las malignas. La naturaleza multifacética de esta enfermedad, que abarca tanto las variantes hialino vascular como de células plasmáticas, subraya la importancia de un examen minucioso de los hallazgos histopatológicos para brindar un diagnóstico oportuno y certero.
DESCRIPCIÓN DEL CASO
Se describe el caso de una paciente de sexo femenino, de 30 años de edad, que no presentaba síntomas evidentes. La paciente acudió a un examen físico rutinario, el cual incluía una evaluación mamaria, durante el mismo se detectó la presencia de una adenopatía en la región supraclavicular izquierda, además de adenopatías en la región axilar ipsilateral. Como parte de la evaluación y para obtener una mejor comprensión de la situación, se indicaron estudios de imagen, consistentes en tomografías contrastadas del cuello, tórax y abdomen. Estos estudios radiológicos proporcionan imágenes detalladas y precisas de estas áreas para ayudar en la identificación de cualquier anomalía o hallazgo significativo.
Reporte tomografía contrastada de cuello tórax y abdomen (hallazgos positivos).
Conglomerado ganglionar en nivel V izquierdo sugestivo de proceso linfoproliferativo, adenomegalias en cuello bilaterales de origen por determinar.
Además, se realizó una biometría hemática que arrojó los siguientes resultados: el recuento de leucocitos fue de 7,360 (con un porcentaje de linfocitos del 20.2%, eosinófilos del 3.1% y neutrófilos del 61.3%). Los valores para la hemoglobina, hematocrito y plaquetas fueron de 14.1 g/dL, 43.7%, y 468,000 respectivamente. Se observó leve trombocitosis, sin encontrar ningún otro hallazgo positivo que indicara alguna anormalidad significativa.

Luego de identificar el hallazgo en las imágenes, se procedió a realizar la resección de la adenopatía supraclavicular izquierda, la cual fue enviada al servicio de anatomía patológica. En donde se recibe tejido ovoide de 4 × 3 × 1.5 cm con peso de 11 g, la superficie es lisa color café grisáceo homogéneo, semifirme, se realiza corte longitudinal donde se evidencia superficie de corte lisa, blanca homogénea.
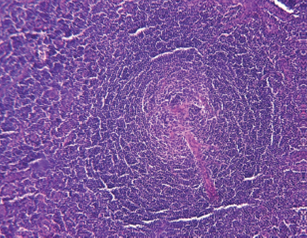
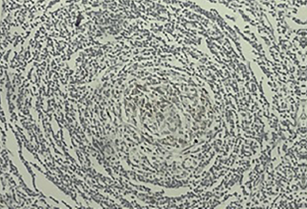
Al examinar el tejido bajo el microscopio, se apreció una proliferación difusa tanto en la corteza como en la médula de varios folículos linfoides agrandados (figura 1). Además, se identificó una proliferación en “capas de cebolla” de la zona del manto, junto con tejido hialino en el centro germinal (figura 2). También se notó la presencia de vasos sanguíneos que mostraban hialinización en los centros germinales (figuras 3 y 4).

Figura 1 Hiperplasia folicular reactiva, con presencia de folículos secundarios que muestran cambios hialinos en centros germinales, proliferación vascular con cambios de hialinización en áreas interfoliculares, fusión de folículos linfoides evidentes en el inferior de la fotomicrografía (H&E) 4X
Imágenes: Vergara Bolaño et al.
Figura 2 Folículo linfoide con disposición de linfocitos en piel de cebolla en zona del manto, además se observa perforado por un vaso sanguíneo con cambios parciales de hialinización imagen clásica lollipop o paleta de caramelo (H&E) 10X
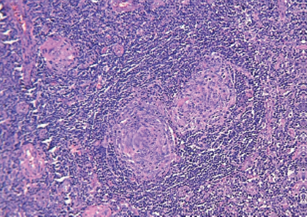
Figura 3 Presencia de más de un centro germinal, con cambios de hialinización en dichos centros (H&E) 10X

Figura 4 Hialinización en vaso sanguíneo localizado en centro germinal, se evidencia el patrón “en piel de cebolla” en la periferia (H&E) 40X
Se realizaron estudios de inmunohistoquímica, en los cuales se observó que CD10 (figura 5 ) mostraba positividad de manera focal en la membrana de las células residuales del centro germinal, mientras que CD20 (figura 6) presentaba positividad de forma difusa en la membrana. Adicionalmente, las pruebas para CD3, CD138, HHV-8 y LMP1 arrojaron resultados negativos.

Basados en los hallazgos histopatológicos y los resultados de los estudios de inmunohistoquímica, se emitió un diagnóstico de enfermedad de Castleman de tipo hialino vascular. Estos hallazgos respaldan la naturaleza de la afección y brindan una base sólida para la caracterización y el entendimiento de esta rara enfermedad en el caso de la paciente.
DISCUSIÓN
La enfermedad de Castleman es un desorden linfoproliferativo poco común, descrito por primera vez en 1956 por el médico patólogo estadounidense Benjamín Castleman en el estudio de pacientes con linfadenopatías mediastinales2. Hasta el momento, no existen datos precisos sobre su incidencia, aunque se estima que hay entre treinta mil y cien mil casos en Estados Unidos3. Esta enfermedad se caracteriza por un agrandamiento de los ganglios linfáticos, qué según su espectro clínico puede ser unicéntrico (limitado a una región) o multicéntrico, para su diagnóstico definitivo requiere el estudio anatomopatológico4.
En cuanto a la presentación del cuadro, puede ser asintomática, principalmente en la variante unicéntrica, donde suele ser captada como un hallazgo incidental durante el examen físico o con la realización de estudios de imagen en los que se encuentren adenomegalias. Normalmente, estas adenopatías presentan un tamaño de 5.5 cm de diámetro5. Los sitios comunes de presentación incluyen tórax (29%), cuello (23%), abdomen (21%) y retroperitoneo (17%); también pueden verse involucrados ganglios linfáticos axilares e inguinales6.
A diferencia de la variante multicéntrica, en donde pueden presentarse síntomas como fiebre, diaforesis, pérdida de peso y hepato-esplenomegalia7 . Es importante tener en cuenta que los síntomas mencionados son inespecíficos y se presentan en múltiples síndromes clínicos, por lo que en ocasiones pueden no estar presentes.
Histológicamente la enfermedad de Castleman se divide en tres variantes: a) hialino vascular (siendo esta la más frecuente, representa del 76-91% de los casos de enfermedad de Castleman unicéntrica)8, b) de células plasmáticas, c) mixta; además de dos variantes clínicas unicéntrica y multicéntrica (mencionadas previamente) según la localización de los ganglios afectados9. En el caso referido en este artículo se evidenció una variante clínica multicéntrica; dada la presencia de adenopatías supraclaviculares, axilares, cervicales, asociada a plexopatía braquial izquierda desarrollada posterior a la resección quirúrgica de la adenopatía enviada a estudio, esta manifestación es de gran relevancia, puesto que la presencia o ausencia de la neuropatía periférica influye en la clasificación y abordaje terapéutico de la enfermedad.

En el estudio de pacientes con enfermedad de Castleman multicéntrica es necesario hacer distinción de si presentan o no relación con el HHV-8 y a su vez determinar si presentan reactividad al virus de inmunodeficiencia humana (VIH), si no es este el caso, se clasificará como una enfermedad de Castleman multicéntrica idiopática9 (figura 7 ) en donde se ha descrito la relación con dos síndromes (síndrome de POEMS, Síndrome de TAFRO).
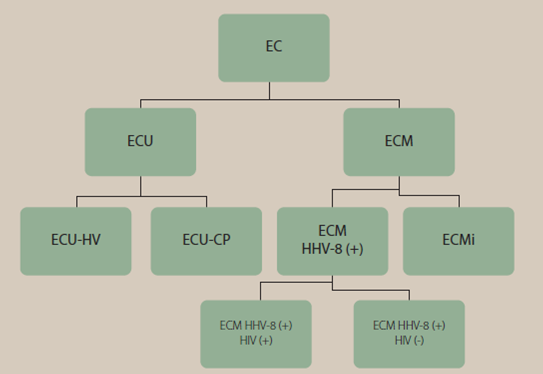
Figura 7 Clasificación enfermedad Castleman según presentación clínica. EC enfermedad de Castleman ECU enfermedad de Castleman unicéntrica ECU -HV variante hialino vascular ECU-CP variante células plasmáticas ECM enfermedad de Castleman multicéntrica ECMi enfermedad de Castleman multicéntrica idiopática, modificado de: Sevilla-Lizcano D, Frías-Soria C, Ortiz-Hidalgo C. Enfermedad de Castleman. Análisis histopatológico e inmunohistoquímico de 39 casos Gaceta medica de México. 2017;153.9
El síndrome de POEMS (acrónimo de polineuropatía, organomegalia, endocrinopatía, gammapatía monoclonal y cambios cutáneos)10 también llamado mielofibroma osteoesclerótico, síndrome Crow-Fukase o síndrome de Taktsuki11 fue descrito por primera vez en 1938, por Scheinker12. En su diagnóstico, se requieren dos criterios principales: la presencia de polirradiculoneuropatía y un desorden monoclonal de células plasmáticas13. Sin embargo, cabe destacar que, dentro de los criterios diagnósticos más amplios para esta enfermedad, también se incluye la enfermedad de Castleman. La relación entre el síndrome de POEMS y la enfermedad de Castleman ya se ha descrito14, de hecho, agrega un componente en la evaluación, diagnóstico y pronóstico de este síndrome complejo15.
Es determinante su identificación en el desarrollo y evolución de la enfermedad, ya que presentarán alteraciones motoras, además de un patrón de poliradiculoneuropatía16.
Respecto al síndrome de TAFRO (acrónimo de trombocitopenia, anasarca, fiebre, mielofibrosis, organomegalia17), descrito por primera vez en el año 2010, en Japón, es una entidad poco frecuente con una incidencia anual de 0.9-4.2 de enfermos por cada millón de habitantes18, histológicamente va a semejar una enfermedad de Castleman multicéntrica en el estudio de los ganglios linfáticos, por lo que se ha considerado por diversos autores como una variante de la enfermedad de Castleman multicéntrica idiopática19, aunque en contraparte otros autores indican la importancia del conocimiento de la etiología del síndrome de TAFRO y así poder realizar una distinción específica con la enfermedad de Castleman multicéntrica20.
Hablando del abordaje en anatomía patológica, se deben identificar inicialmente aspectos macroscópicos. Entre ellos, se observa un agrandamiento difuso de los ganglios afectados, que varía en tamaño desde 2 hasta 20 centímetros. Los mismos van a tener una consistencia firme y una superficie de corte blanca lisa, la cual puede asociarse a calcificaciones21 histopatológicamente, según los hallazgos se divide en variante hialino vascular o de células plasmáticas.
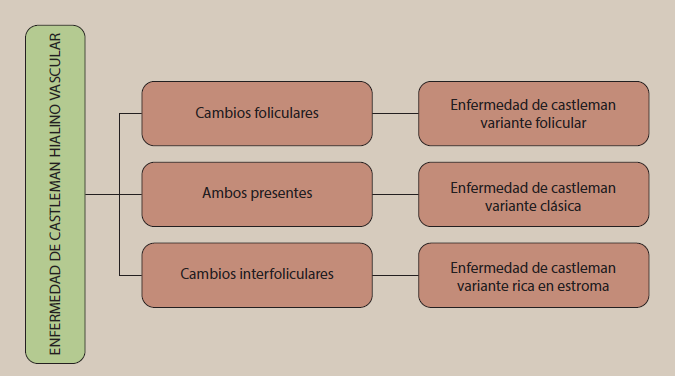
Con respecto a la variante hialino vascular, se pueden dividir los cambios en tres grupos, foliculares, interfoliculares o mixtos22(figura 8).
Características histológicas:
Arquitectura conservada con cierto grado de distorsión.
Folículos linfoides distribuidos por toda la corteza y la médula.
Pueden tener más de un centro germinal.
Presencia de células dendríticas foliculares en los centros germinales.
Depósitos hialinos y vasos sanguíneos escleróticos penetran los centros germinales (lesión hialino vascular).
Patrón en “piel de cebolla” por la formación de anillos concéntricos de linfocitos.
En la variante de células plasmáticas histológicamente encontramos las siguientes características:
La arquitectura de los nódulos linfáticos está preservada.
Plasmocitosis marcada en áreas interfoliculares de ahí su nombre.
Vasculatura prominente en áreas interfoliculares.
Entre folículos linfoides se puede encontrar mayor espacio, centros germinales hiperplásicos.
Zonas del manto bien definidas y pueden expandirse23.
Por otro lado, en el patrón inmunohistoquímico se identifican rasgos de expresión dependiendo de las características clínicas de la enfermedad (uni-multicéntrico) y según la clasificación histológica enfermedad de Castleman hialino vascular (EC HV) o enfermedad de Castleman de células plasmáticas (EC CP) (tabla 1).
Tabla 1 Perfil inmunohistoquímico variantes enfermedad Castleman
| EC HV | EC CP |
|---|---|
| Células dendríticas foliculares de los anillos concéntricos positivas para CD21, CD23, CD35, EGFR, VCAM1 | Células plasmáticas en las áreas interfoliculares (+) CD138 |
| Células residuales en los centros germinales positivas para CD10 BCL6 | CP con perfil HHV-8 negativo suelen ser positivos CD68 |
| En las zonas del manto positivos para CD19, CD20, BCL2 | CP con perfil HHV-8 positivo suelen ser positivos para LNA-1 IL-6 viral |
EC HV: enfermedad de Castleman variante hialino vascular; EC CP: enfermedad de Castleman variante células plasmáticas.
Al considerar las características histopatológicas y el perfil inmunohistoquímico, es fundamental tener en mente los principales diagnósticos diferenciales. Entre estos se incluyen trastornos benignos como la enfermedad de Kimura. La enfermedad de Kimura, de etiología desconocida24, fue descrita por primera vez en 1937 como una forma de linfogranuloma hiperplásico eosinofílico. Sin embargo25, su caracterización completa se logró en 1948, por Kimura e Ishikawa, de ahí su denominación. A pesar de las notables diferencias epidemiológicas en comparación con la enfermedad de Castleman, dado que la enfermedad de Kimura tiende a manifestarse principalmente en países orientales y se caracteriza por la presencia de lesiones indoloras localizadas principalmente en cabeza y cuello26, principalmente subcutáneas, en glándulas salivales o ganglios linfáticos cervicales27, se pueden identificar similitudes histológicas con la enfermedad de Castleman.
En ambos casos se observa hiperplasia de folículos linfoides junto con una proliferación vascular en los centros germinales, así como la presencia de células plasmáticas. Sin embargo, existen diferencias distintivas entre la enfermedad de Kimura y la enfermedad de Castleman. Por ejemplo, en la enfermedad de Kimura, es posible identificar eosinófilos en los centros germinales, una característica que no es común en la enfermedad de Castleman. Además, en la enfermedad de Kimura, no se presentan los depósitos hialinos ni las características en forma de cebolla (lollipop)28 mencionadas e ilustradas previamente en la enfermedad de Castleman.
Otra entidad benigna que merece mención es la linfadenopatía reactiva, esta junto con la enfermedad de Castleman son dos afecciones que afectan los ganglios linfáticos, pero se distinguen por sus características clínicas, histológicas y etiológicas. La linfadenopatía reactiva, generalmente desencadenada por infecciones (principalmente virales y siendo más frecuentes en la población infantil)29 o respuestas inflamatorias, se manifiesta con ganglios linfáticos más pequeños, móviles y tiende a ser bilateral. Histológicamente, muestra expansión de la paracorteza y centro germinal, así como un aumento de células plasmáticas.
Por otro lado, la enfermedad de Castleman, un trastorno linfoproliferativo poco común, presenta síntomas sistémicos como fiebre, fatiga y pérdida de peso sobre todo en la variante multicéntrica, además de afectar los ganglios linfáticos. Estos ganglios pueden variar en tamaño y distribución, pudiendo ser unicéntricos o multicéntricos. Histológicamente, se caracteriza por una arquitectura ganglionar aumentada con hiperplasia linfoide, células plasmáticas y, en algunos casos, depósitos hialinos. Estas características histopatológicas son fundamentales para establecer un diagnóstico diferencial.
En resumen, la linfadenopatía reactiva se asocia con respuestas inflamatorias a infecciones y muestra ganglios linfáticos más pequeños, mientras que la enfermedad de Castleman es un trastorno linfoproliferativo con síntomas sistémicos, variaciones en el tamaño y una histología distintiva. Comprender estas diferencias es esencial para un diagnóstico y manejo adecuado de estas condiciones.
Uno de los principales diagnósticos diferenciales se encuentra en los pacientes con infección por VIH, en donde, en casos avanzados de la infección por VIH, pueden aparecer manifestaciones similares a las observadas en la enfermedad de Castleman, incluyendo características vasculares y fenómenos de hialinización en los ganglios linfáticos. Para establecer un diagnóstico diferencial definitivo, es esencial acceder al expediente clínico del paciente y realizar estudios serológicos detallados. Estos registros médicos y análisis específicos pueden proporcionar información crítica para distinguir entre ambas afecciones y garantizar un enfoque adecuado en el diagnóstico y tratamiento de los pacientes con estas manifestaciones clínicas.
En relación a los principales diagnósticos diferenciales malignos, el plasmocitoma se incluye en esta categoría. Aunque la presentación de plasmocitomas primarios en los ganglios linfáticos es poco común, suele ser característica de tumores primarios que han metastatizado a los ganglios linfáticos30.
Las diferencias histológicas entre el plasmocitoma y la enfermedad de Castleman son notables. El plasmocitoma se caracteriza por una proliferación localizada de células plasmáticas malignas31 en el tejido afectado, con una presencia dominante de células plasmáticas atípicas que a menudo muestran depósitos intracelulares y extracelulares de inmunoglobulinas monoclonales. Esta entidad tiende a presentar una estructura más homogénea en comparación con la enfermedad de Castleman.
En contraste, la enfermedad de Castleman se caracteriza por una arquitectura ganglionar aumentada con hiperplasia linfoide y una marcada proliferación vascular en los centros germinales de los ganglios linfáticos. Si bien también puede incluir células plasmáticas, su cantidad y distribución varían y pueden estar acompañadas por depósitos hialinos, una característica distintiva de esta enfermedad. Esta variabilidad histológica puede reflejar las diferentes subtipos y presentaciones clínicas de la enfermedad de Castleman.
En resumen, mientras que el plasmocitoma se centra en la proliferación localizada de células plasmáticas malignas con depósitos de inmunoglobulinas, la enfermedad de Castleman se caracteriza por la hiperplasia ganglionar, la proliferación vascular y una variabilidad histológica que puede incluir células plasmáticas, depósitos hialinos y una arquitectura ganglionar compleja.
En la continuación del análisis diferencial de la enfermedad de Castleman, es importante considerar la posibilidad de linfoma. En relación a este tipo de cáncer que se origina en el sistema linfático, las líneas mayormente afectadas son las de linfocitos B y T, las cuales desempeñan un rol crucial en la inmunidad adaptativa, tanto en la respuesta celular como en la humoral, respectivamente32. El linfoma se clasifica en dos tipos principales: linfoma no Hodgkin (que representa el 90% de la totalidad de los casos de linfoma) y linfoma de Hodgkin (el 10% restante)33. Cada uno de estos tipos presentará subclasificaciones dependiendo de la línea celular afectada.
El linfoma folicular representa el 20% de todos los linfomas34, incluso es el más frecuente de los linfomas indolentes35, forma parte de los linfomas de células B; en este se ha identificado la translocación t(14;18) en aproximadamente el 85% de los casos, lo que llevará a una sobreexpresión de BCL-236, histológicamente el linfoma folicular reproduce o imita los centros germinales normales de los folículos linfoides secundarios, según la clasificación de la OMS se divide en tres grados, dependiendo del número de centroblastos por campo de alto poder, el patrón morfológico y el perfil inmunohistoquímico que presenten. Los folículos linfoides atípicos están compuestos por células de tamaño pequeño/mediano (centrocitos) y otras de mayor tamaño (centroblastos)37. En el linfoma folicular de bajo grado se pueden presentar similitudes con la variante hialino vascular de la enfermedad de Castleman como son el patrón folicular y la proliferación vascular. Sin embargo, existen diferencias distintivas que permiten discernir entre ambas condiciones. En la enfermedad de Castleman, se observará una proliferación de la zona del manto, que puede estar atenuada o incluso ausente en el linfoma folicular. Además, en la enfermedad de Castleman, los folículos linfoides estarán más espaciados en comparación con los del linfoma folicular. En lo que respecta al inmunofenotipo, tanto en el linfoma folicular como en la enfermedad de Castleman, observaremos positividad de marcadores B, como CD19 y CD20. Sin embargo, en el linfoma folicular, encontraremos una positividad difusa en las células tumorales para CD10 y BCL638. Estos últimos marcadores se expresan de manera focal en la enfermedad de Castleman.
Dentro del contexto del linfoma de células del manto, el cual forma parte de los linfomas de células B e incluso se incluye entre los linfomas indolentes39, representa aproximadamente el 7% de los linfomas no hodgkin40 junto al linfoma folicular, se erigen como los principales diagnósticos diferenciales en términos de entidades malignas, basados en las características morfológicas de la enfermedad de Castleman. En este contexto, se enfatiza la prominencia de los folículos y la formación de anillos concéntricos por parte de los linfocitos en la zona del manto, aspectos que comparten con la enfermedad de Castleman. Por lo tanto, el papel esencial del inmunofenotipo en el proceso de diagnóstico diferencial se vuelve evidente.
El perfil inmunofenotípico característico del linfoma de células del manto abarca la positividad para CD5, ciclina D1 y SOX1141. En contraste, CD10 tiende a presentar negatividad en este tipo de linfoma, aunque en situaciones atípicas con variaciones en el inmunofenotipo podría mostrar positividad42. Estos marcadores inmunohistoquímicos divergen de los encontrados en la enfermedad de Castleman. Es importante destacar en el linfoma de las células de manto la importancia, y obligatoriedad de la solicitud y reporte del porcentaje de replicación de las células tumorales representado mediante el marcador Ki-67 ya que es tomado como factor pronóstico de la entidad43.
En resumen, aunque los linfomas foliculares y de células del manto pueden presentar características similares a la variante hialino vascular de la enfermedad de Castleman, un análisis detallado de los patrones de proliferación de los folículos, así como el perfil inmunofenotípico, resultan esenciales para llevar a cabo un diagnóstico diferencial preciso entre estas entidades clínicas.
CONCLUSIÓN
En este artículo se presentó el caso de una paciente de 30 años con diagnóstico de enfermedad de Castleman, una entidad poco común y heterogénea que puede presentarse con una amplia variedad de características clínicas y patológicas.
La enfermedad de Castleman es un trastorno del sistema linfático que se caracteriza por la proliferación anormal de células linfoides en los ganglios linfáticos o tejidos linfoides extra ganglionares. En el caso de esta paciente, se identificaron características histológicas como son el patrón en piel de cebolla, formado por linfocitos con distribución concéntrica en la zona del manto, la fusión de centros germinales, el patrón de paleta de dulce lollipop formado por la presencia de un vaso sanguíneo hialinizado en el centro germinal, que apoyaron el diagnóstico de enfermedad de Castleman.
Las características histológicas de la enfermedad de Castleman pueden variar y se clasifican en tres subtipos principales: hialino-vascular, mixto y de células plasmáticas. En este caso, se observaron hallazgos histológicos consistentes con el subtipo hialino-vascular, que se caracteriza por la presencia de vasos sanguíneos dilatados y con cambios hialinos, rodeados de una proliferación de células linfoides.
Es importante destacar que el diagnóstico diferencial de la enfermedad de Castleman incluye otras enfermedades linfoproliferativas y neoplasias, como es el linfoma no Hodgkin, principalmente sus subtipos de células del manto y folicular. Estos diagnósticos diferenciales deben ser considerados y excluidos mediante estudios adicionales, como inmunohistoquímica y técnicas moleculares, para confirmar el diagnóstico final.
En resumen, el diagnóstico de enfermedad de Castleman requiere una evaluación exhaustiva de las características histológicas y la exclusión de otros diagnósticos diferenciales. La identificación de los subtipos histológicos puede ayudar a guiar el manejo y el pronóstico de los pacientes. Un enfoque multidisciplinario es esencial para garantizar un diagnóstico preciso y un manejo adecuado de estos pacientes.

