











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Las demencias rápidamente progresivas son entidades clínicas de rápida instauración que se manifiestan con un deterioro cognitivo agudo, el cual puede atribuirse a un cuadro multifactorial que presenta un subgrupo de etiologías potencialmente reversibles con tratamiento precoz (tabla 1). Para fines prácticos podemos definirla como la instauración de la demencia a partir de un periodo de 2 años del inicio de los síntomas1.
Tabla 1 Etiología de demencias rápidamente progresiva (VITAMINS)
| Etiología subyacente | Ejemplos |
| Vascular | |
| Infecciosa | |
| Tóxica/metabólica-alcohólica | |
| Autoinmune | |
| Metástasis/neoplásicas | |
| Iatrogénicas | |
| Neurodegenerativas | |
| Sistémicas/endocrinas/estructurales |
Actualmente en el área neuropsiquiatría el término demencia ha evolucionado a: trastorno neurocognitivo mayor un término global que describe un conjunto amplio de síntomas asociados a un deterioro mental (memoria, pensamiento), lo bastante grave como para reducir la capacidad de la persona para realizar las actividades diarias (tabla 2) 2.
Tabla 2 Criterios diagnósticos de demencia rápidamente progresiva
| A. Evidencias de un declive cognitivo significativo comparado
con el nivel previo de rendimiento en uno o más dominios cognitivos (atención compleja, función ejecutiva, aprendizaje y memoria, lenguaje, habilidad perceptual motora o cognición social) basadas en: |
| B. Los déficits cognitivos interfieren con la autonomía del
individuo en las actividades cotidianas (es decir, por
lo menos necesita asistencia con las actividades instrumentales complejas de la vida diaria, como pagar facturas o cumplir los tratamientos). |
| C. Los déficits cognitivos no ocurren exclusivamente en el contexto de un delirium. |
| D. Los déficits cognitivos no se explican mejor por otro trastorno mental (p. ej., trastorno depresivo mayor, esquizofrenia). |
Entre ellas está la enfermedad de Creutzfeldt-Jakob (ECJ) es una entidad neurodegenerativa, neuroselectiva y fatal con tiempo de incubación prolongada, que se debe sospechar en todo paciente que presente una demencia rápidamente progresiva, asociada a otros datos de afección de áreas de sistema nervioso central3,4.Es producida por la acumulación de una proteína celular denominada proteína priónica, prion resistente a proteasas (PrPRES), la cual sufre una alteración en su estructura secundaria en un proceso de postraducción generando partículas infecciosas4-6. El cuadro clínico de esta enfermedad se caracteriza por presentarse como una demencia rápidamente progresiva, acompañada comúnmente de mioclonías y ataxia. Se observan igualmente manifestaciones piramidales, extrapiramidales e incluso ceguera occipital. Posteriormente se observa una fase de mutismo acinético que se extiende hasta el fallecimiento del paciente7.Presenta una incidencia anual de un caso por millón de personas8. Corresponde a la forma más común de las encefalopatías espongiformes transmisibles, también conocidas como enfermedades priónicas9. Presenta un patrón histopatológico caracterizado por pérdida de cuerpos neuronales, proliferación de células gliales, y aparición de vacuolas en el citoplasma de la neurona, que le dan la apariencia de una esponja, de ahí que pertenezca al grupo de enfermedades llamadas encefalopatías espongiformes. Existen 3 formas de enfermedades priónicas: hereditaria, adquirida o esporádica. Esta última, de acuerdo con sus características clínicas y patológicas, se divide en ECJ, insomnio familiar fatal y el Kuru10. La ECJ se considera la encefalopatía espongiforme transmisible más común en humanos, siendo el 85% de los casos reportados de tipo esporádico, mientras que un 15% restante lo consisten la forma familiar de la enfermedad y la forma de transmisión iatrogénica11. Su inicio es por lo general entre los 45 y 75 años, con una edad media de inicio de 60 años12. El diagnóstico preciso de la enfermedad priónica depende de un cuidadoso examen clínico y de la interpretación de pruebas diagnósticas, que incluyen electroencefalograma, marcadores como la proteína 14-3-3, proteína Tau y neuroimágenes13,14.
REPORTE DE CASO
Paciente del sexo masculino de 40 años, que cuenta con antecedentes de vivienda en Washington, EE. UU., al inicio del padecimiento, 4 meses antes de recibir manejo neurológico. Inició su padecimiento al presentar de manera insidiosa alteración de memoria a corto plazo, por lo que se comenzó tratamiento psiquiátrico no especificado durante 4 meses, sin mejoría; posterior a ello acudió a valoración neurológica donde se realizó una prueba MOCA donde obtuvo 16/30, se inició tratamiento con memantina, el cual no evidenció cambios.
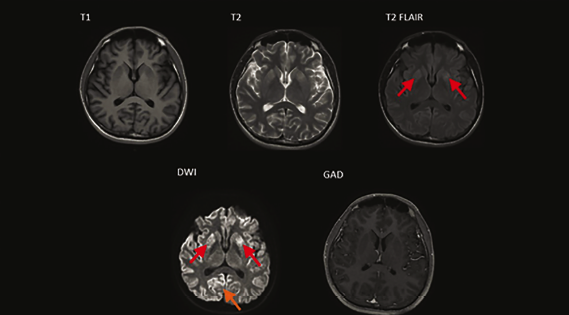
La sintomatología progresó con episodios depresivos y labilidad emocional con tendencia a la irritabilidad, 3 semanas después se agregó desorientación, disminución de fuerza en hemicuerpo izquierdo y latero pulsión de la marcha ipsilateral. Fue ingresado a hospitalización donde se realizaron distintas pruebas entre las cuales destacaron como positivas la proteína 14-3-3, proteína TAU 15,134 pg/ml (normal 0-1150 pg/ml), y resonancia magnética, la cual presentó “listón cortical”, e hiperintensidades en los núcleos de la base (figura 1), a su vez cabe destacar que no hubo mutaciones para el gen PRNP, pero presentó una mutación silenciosa en el codón 117 y polimorfismo en Val en codón 129.
Imágenes: Guevaraet al.
Figura 1. En las imágenes de resonancia magnética en cortes axiales donde se observan cambios en los núcleos de la base en las secuencias potenciadas en T2 donde se observa restricción simétrica en la cabeza del núcleo caudado (flecha roja) y restricción a la difusión en DWI asimétrico cortical, signo del listón cortical (flecha naranja).
Se le dio el alta con valproato de magnesio; 15 meses después del inicio de la sintomatología neurológica fue ingresado por presentar alzas térmicas, dificultad respiratoria y tos productiva, por lo cual fue ingresado al nosocomio cumpliendo criterios de neumonía adquirida en la comunidad, la cual fue tratada con un triple esquema antibiótico, sin mejoría. En el hospital se le realizó una tomografía, la cual fue compatible con hemotórax y un absceso pulmonar tabicado, por lo que se dio manejo antibiótico y se drenó. Durante su estancia, el paciente perdió aproximadamente 5 kilos y presentó incapacidad para alimentarse, por lo que se utilizó nutrición parenteral ante la dificultad de llevar a cabo el procedimiento quirúrgico por la enfermedad priónica. El paciente permaneció 25 días hospitalizado hasta fallecer de sepsis con origen pulmonar. No fue realizada necropsia por falta de condiciones de seguridad adecuadas por parte del servicio de patología.
DISCUSIÓN
La ECJ pertenece a una pequeña, rara y fatal familia de desórdenes degenerativos, que se relaciona con una alteración de una proteína priónica intracelular (PrP). Dada la morbimortalidad asociada a esta patología y al alto riesgo de transmisión, el diagnóstico oportuno de esta enfermedad es vital. Sin embargo, actualmente los criterios son controversiales y no hay un consenso definitivo sobre su diagnóstico, el cual se hace usualmente en las fases terminales o post mortem, por lo que es necesario tomar ciertos rubros clínicos, epidemiológicos, genéticos, citoquímicos y de imagen para llegar a un diagnóstico que será principalmente de exclusión.
El diagnóstico definitivo antes se consideraba exclusivamente patológico, actualmente debido a lo peligroso de la biopsia o necropsia es posible realizarlo tomando en cuenta los signos clínicos como ataxia cerebelosa, demencia rápidamente progresiva y mioclonus, además de una serie de estudios claves como resonancia magnética, análisis del líquido cefalorraquídeo en donde se realiza búsqueda de proteína 14-3-3 y proteína Tau, así como apoyarse en ciertos hallazgos en la resonancia magnética, esto con el fin de integrar el diagnóstico de forma clásica según los criterios de la OMS1,15(tabla 3).
Tabla 3 Criterios diagnósticos de Enfermedad de Creutzfeldt-Jakob, OMS 1998
| A. Demencia progresiva |
| B. Manifestaciones neurológicas específicas |
| C. Pruebas de Laboratorio |
| D. Los tests complementarios no deben sugerir otra patología. |
| Probable = A + Al menos 2 de B + Al menos 1 de C. Posible = A + Al menos 2 de B + duración menor a 2 años. |
La proteína 14-3-3 es una enolasa citosólica, cuya presencia indica daño cerebral producto de muerte neuronal, tiene una sensibilidad de 92% y una especificidad de 80%15. Si bien la detección de la proteína 14-3-3 no genera un diagnóstico confirmatorio de la ECJ, se encuentra presente solo en un bajo porcentaje de otros cuadros a plantear como diagnósticos diferenciales, nosotros optamos por solicitarla principalmente por la rápida progresión de los síntomas, al ser parte de un escrutinio en este tipo de demencias.
En este reporte de caso se presenta un paciente que inició con un cuadro inespecífico de alteración de memoria, el cual fue progresando paulatinamente, recibió atención médica por diversos especialistas como psiquiatra y neurólogo, donde no hubo mejoría alguna tras diversos tratamientos, los síntomas que nos orientaron a pensar en esta patología como diagnóstico diferencial fueron los episodios depresivos, labilidad emocional con tendencia a la irritabilidad, así como la desorientación, disminución de fuerza en hemicuerpo izquierdo y lateropulsión de la marcha ipsilateral, ocurriendo esto de manera progresiva en un periodo relativamente corto.
Respecto a la clínica de inicio, esta puede ser muy variada; en este caso, se presentó de manera inespecífica, con sintomatología general, acompañada de un trastorno depresivo, y alteraciones del estado de ánimo. Este tipo de presentación no es infrecuente, encontrándose que alrededor del 35% de los pacientes debutan con síntomas consistentes en fatiga, depresión, trastornos del sueño, pérdida de peso y apetito antes de la aparición del cuadro neurológico. Por lo anterior, no es raro que estos pacientes sean estudiados por diferentes especialistas (psiquiatra, internista), antes de llegar al neurólogo16.
Ante el agravamiento de los síntomas, el paciente se decidió internar para realizar una amplia batería de pruebas diagnósticas en las cuales mediante estudio de líquido cefalorraquídeo se descartan enfermedades infecciosas mediante cultivos y tinciones negativos.
Respecto al electroencefalograma, destacó la presencia de ondas lentas durante todo el registro y 2 crisis mioclónicas. El seguimiento electroencefalográfico es esencial para lograr un mejor acercamiento etiológico del deterioro cognitivo subagudo, puesto que el electroencefalograma (EEG) puede cambiar en el curso del cuadro clínico y en algunos casos el patrón clásico aparece solo tardíamente. Por otro lado, hay que considerar también que el patrón clásico de ondas periódicas de morfología trifásica de frecuencia 1.5-2 cps es idéntico al que se ve en muchas otras encefalopatías, por lo cual su especificidad se aplica solo cuando existe una historia clínica sugerente18.
Dentro de este panorama clínico se deben hacer diagnósticos diferenciales con otras demencias progresivas, por lo cual se solicitaron diversos anticuerpos como anti-NMDA, anti GAD, anti-HU, anti-Ri, los cuales fueron negativos, en este caso los síntomas neuropsiquiátricos que más nos orientaron a este diagnóstico fueron los episodios depresivos, la irritabilidad y las alteraciones de lateralización que darían inicio al cuadro demencial1.
La resonancia magnética es otra herramienta importante en el diagnóstico diferencial de una demencia rápidamente progresiva y tiene una alta sensibilidad y especificidad en el diagnóstico de ECJ, nuestro paciente presentó hiperintensidades en los núcleos de la base y encorvamiento cortical, hallazgos incluidos en los criterios de resonancia magnética para ECJ de la universidad de San Francisco 2010. Para integrar estos criterios usamos el consenso de la universidad para la probabilidad de la enfermedad, siempre descartando otras patologías siguiendo el algoritmo VITAMINS propuesto por Geschwind2,16.
Mediante el estudio genético fue posible la secuenciación del gen PRNP donde no se evidenciaron mutaciones, lo cual permite descartar una forma familiar de la enfermedad. La evidencia de mutación silenciosa del gen 117 y el polimorfismo para valina del codón 129 presentes en el paciente, se han asociado a formas adquiridas y esporádicas de la enfermedad17.
En cuanto a la cronología, el paciente tuvo una evolución desde el inicio de los síntomas hasta su fallecimiento de 18 meses, ingresando con neumonía 15 meses después del inicio sintomático neurológico, por lo cual se puede catalogar como neumonía adquirida en la comunidad, los trastornos neuropsiquiátricos fueron causa de incapacidad importante a partir de 3 meses, al comienzo del cuadro, la cual le generó una dependencia total aproximadamente un mes antes de su fallecimiento. Es relevante resaltar el tiempo de demora para abordaje neurológico durante la evaluación psiquiátrica, común en este tipo de patologías por su comienzo insidioso con cambios en el estado de ánimo poco específicos.
El paciente no refirió antecedentes quirúrgicos ni transfusionales los cuales puedan intuir hacia una forma adquirida, así mismo se puede descartar la forma familiar por el estudio genético, lo cual nos hace concluir que se trata de una forma esporádica11.
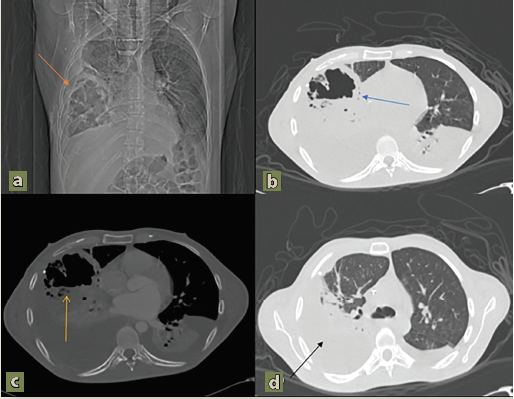
Durante su ingreso recibió tratamiento para el absceso pulmonar (figura 2) a base de meropenem, el cual fue secundario a la neumonía que presento, esto atribuible a su condición neurológica, así mismo recibieron manejo las complicaciones asociadas como desequilibrio hidroelectrolítico, permaneciendo en el nosocomio 17 días hasta su fallecimiento.
Imágenes: Guevara et al.
Figura 2 a, b, c) La arquitectura del parénquima pulmonar se encuentra alterada a expensas de imagen que se localiza el lóbulo medio e inferior del pulmón derecho, de características ovoide, paredes engrosadas, irregulares, con interior líquido-aire, que realza de forma difusa tras la administración de medio de contraste, mide aproximadamente 8.3 x 7.7 x 8.2 cm en sus ejes mayores para un volumen aproximado de 274.0 cc, por imagen compatible con parénquima destruido por absceso y presencia de una atelectasia posterobasal izquierda. d) Derrame pleural bilateral de predominio derecho aproximadamente de 50% de este lado con colapso pulmonar pasivo secundario y aspecto lobulado/septado altamente sospechoso de empiema.
Usualmente la biopsia ha sido considerada el gold standard para el diagnóstico de ECJ, pero en la actualidad se está optando por no realizarla debido a que su utilización no siempre es diagnóstica, llegando a demostrar tasas de éxito de solo el 57% e incluso su positividad no altera el tratamiento del paciente, pero en cambio sí presenta un riesgo de transmisión, esto debido a que estos priones no son destruidos por formalina o por fijación con formalina fenolizada y son resistentes a los métodos de descontaminación física y química, así mismo estos patógenos pueden ser transmitidos por tejidos previamente fijados en formalina o parafina, incluso se ha reportado infección de un técnico, el cual trabajaba con cerebros incluidos en parafina19,20.

