











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink

INTRODUCCIÓN
La histiocitosis de células de Langerhans es una entidad que puede presentarse como lesiones neoplásicas localizadas o de manera diseminada con afección multiorgánica. Se cree que podría estar relacionada con trastornos inmunológicos y con un oncogén denominado BRAF V600E. La incidencia es mayor en niños, con 0.2 y 2.0 casos por cada 100,000 menores de 15 años de edad, predominando entre los 2 y 4 años de edad, afectando más al género masculino (60-70%). Las lesiones focalizadas representan el 65% de los casos, siendo el tejido óseo el de mayor incidencia con un 90%. Se caracteriza por lesiones osteolíticas en el techo y pared posteroexterna1,2.
El objetivo de esta presentación es exponer un caso de histiocitosis de células de Langerhans en su variedad orbitaria como lesión única, una entidad poco frecuente, sobre todo en pacientes pediátricos: esta última representa el 1-2% de tumores orbitarios.
PRESENTACIÓN DEL CASO
Paciente del sexo femenino de 4 años de edad, quien debutó con proptosis izquierda, diplopía, fiebre y pérdida de la agudeza visual de tres semanas de evolución (figura 1). En la ultrasonografía ocular modo A y B se aprecia, en la órbita izquierda, una lesión extra conal; su reflectividad interna varía entre 23 y 37 mHz, no se aprecia vascularidad. Este último hallazgo dirige el diagnóstico a un linfangioma. En la tomografía computarizada (TAC) se observa una lesión extra axial, hiperdensa, de aproximadamente 3.7 × 2.97 × 2.36 cm en sus ejes mayores, la cual provoca erosión en hueso esfenoidal hacia rostral y hacia la fosa anterior; así como ocupación parcial del seno esfenoidal y desplazamiento anterior de las estructuras de la órbita (figura 2). A la aplicación del medio de contraste se aprecia reforzamiento homogéneo de aproximadamente 40 unidades Hounsfield, sin alteraciones en el parénquima cerebral
Se inició tratamiento con antiinflamatorios, con lo que se disminuyó de forma significativa la proptosis. En tan solo 7 días después del tratamiento médico, la sintomatología descrita previamente recurrió súbitamente.
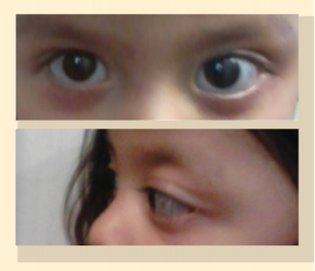
Figura 1 Exploración física inicial de la paciente. a) Proptosis de ojo izquierdo danos en la exoftalmometría ojo derecho 9.5 mm ojo izquierdo 14 mm. Movimientos oculares sin alteraciones, sin limitaciones o inconstancias. b) Ojo izquierdo con retracción palpebral inferior.

Figura 2 Tomografía axial computarizada de cráneo corte coronal. Observe que existe lesión extra axial, hiperdensa, así como ocupación parcial del seno esfenoidal y desplazamiento anterior de las estructuras de la órbita.
En la revaloración se apreció proptosis izquierda y las medidas de exoftalmometría arrojaron los siguientes resultados: 180 mm, 13 mm derecha y 18 mm izquierda, sin tumoración evidente. A la palpación la consistencia era blanda y renitente. Se observó lagoftalmos de 2 mm en ojo izquierdo. Los movimientos oculares se mostraban con restricción a la supra e infraversión. La agudeza visual se mostró fija y con adecuado seguimiento de objetos, a la refracción bajo cicloplegia, +2.00 dioptrías de ojo derecho y +3.00 de ojo izquierdo. La presión intraocular a la dígitopresión se apreciaba elevada en ojo izquierdo. En la córnea izquierda se observó una queratoplastía punteada superficial, ángulos abiertos, pupila normorrefléctica y cristalino transparente sin alteraciones. En el polo posterior se visualizaba el nervio óptico de coloración naranja, una excavación del 30% de ambos ojos y sin alteraciones en la mácula.
Se realizó biopsia excisional en la órbita izquierda por abordaje de craneotomía coronal. Fueron enviadas 2 muestras en fresco para estudio transoperatorio, etiquetándose la primera como “lesión intraorbitaria (TOP)” y la segunda como “lesión orbitaria izquierda”.
Descripción macroscópica
La primera muestra enviada constó de un fragmento rugoso, blanco-grisáceo, blando, irregularmente ovoide y de 0.5 × 0.4 × 0.3 cm. La segunda muestra, de 4.0 × 3.5 cm, se caracterizó por múltiples fragmentos irregulares de tejido. Fue descrita por su aspecto carnoso de color café claro, friables y acompañados de fragmentos óseos.
Descripción microscópica
En los cortes de ambas muestras obtenidas por inclusión en parafina a 4 micras de espesor y teñida con hematoxilina-eosina, se observó una lesión maligna rodeada por células redondas con moderada cantidad de citoplasma claro, los núcleos focalmente en forma de hendiduras, con intenso infiltrado inflamatorio por eosinófilos, con zonas de necrosis y hemorragia.
En los cortes obtenidos en forma transoperatoria se encontraron los mismos elementos descritos, por lo que se reportó como: tumor maligno de células pequeñas redondas y azules, compatible con rabdomiosarcoma. Para confirmar diagnóstico se realizaron pruebas de inmunohistoquímica con diversos marcadores seleccionados, con base en los hallazgos microscópicos. Los anticuerpos que resultaron positivos fueron CD1A (Bio&SB) y S100 (Bio&SB); mientras que resultaron positivos focales lisozima (Bio&SB) y CD68 (Bio&SB) (figura 3). Los hallazgos anteriores, fueron concluyentes para el diagnóstico de histiocitosis de células de Langerhans.
Se comenzó con ciclo de quimioterapia a base de vincristina y ciclofosfamida, una vez al mes durante un año. Durante el control se realizó TAC y gammagrafía ósea, en las que se observa remisión de la enfermedad. Sin embargo, la paciente aún presentaba astenia, adinamia, polidipsia, poliuria e irritabilidad. Debido a esto, fue evaluada por el departamento de endocrinología, el cual realizó el diagnóstico de diabetes insípida por probable infiltración histiocitaria a hipófisis, por lo que se inició tratamiento con desmopresina.

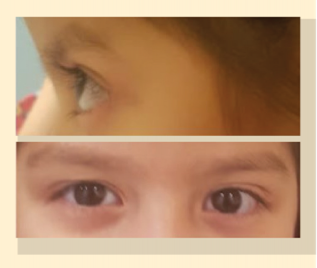
Doce meses después de haber concluido la quimioterapia, se observaron tres tumoraciones en la zona maxilar inferior izquierdo y dos en la porción occipital y parietal izquierda occipital. Se determinó por gammagrafía que solo involucraban tejido óseo. Se inició segunda sesión de quimioterapia con el mismo esquema de tratamiento previamente descrito. A la paciente se le otorgó el alta de oncología con estudios complementarios sin datos de enfermedad activa (figura 4).
DISCUSIÓN
El rabdomiosarcoma es el tumor de órbita infantil maligno más común, el cual se manifiesta por proptosis progresiva, generalmente de rápida evolución. Es un tumor agresivo, el cual frecuentemente se relaciona al desarrollo de metástasis; por lo que su diagnóstico debe ser oportuno con base en su sospecha. La asimetría mayor a 2 mm entre ambos ojos sugiere una proptosis unilateral3. Frente a una exoftalmía unilateral se debe sospechar en primer lugar en un tumor orbitario, la cual puede ser secundaria a una anomalía vascular, a un proceso inflamatorio o masa tumoral. Dependiendo de la rapidez del desarrollo de la proptosis y la inflamación asociada, ésta puede ser o no ser dolorosa. Otros síntomas que pueden acompañar son: irritación ocular, diplopia, estrabismo, disminución de la agudeza visual y lagrimeo. Si la agudeza visual es subnormal, se asume que el nervio óptico o el glóbulo ocular está siendo deformado. Los movimientos oculares reducidos indican inflamación de los músculos extraoculares, lo que amenaza la función del nervio óptico1,2.
Los diagnósticos diferenciales incluyen una variedad de trastornos entre los que se encuentran: lesiones quísticas, celulitis orbitaria, linfoma, pseudotumor inflamatorio, histiocitosis de células de Langerhans o infiltraciones metastásicas de otros tumores, como el neuroblastoma4.
El linfangioma o también conocido como higroma quístico, es un tumor de origen vascular que se cataloga como lesión benigna que puede involucrar a la órbita. Son malformaciones vasculares constituidas por elementos venosos y linfáticos del cuello, axila, mediastino y retro peritoneo. La acumulación de líquido puede ser propensa a procesos infecciosos4. Se clasifica en capilar, cavernoso y quístico, los cuales pueden coexistir, aparece frecuentemente en los primeros 5 años de vida. Clínicamente los linfangiomas se presentan con proptosis, con hemorragia circunscrita intralesional de forma espontánea o después de cuadros gripales, traumatismos con aumento de tamaño y formación de “quistes de chocolate”. Esto ocurre cuando la pared de un vaso sanguíneo es destruida y vierte su contenido dentro de los canales linfáticos5.
Las técnicas diagnósticas de elección son la tomografía computarizada, la resonancia magnética y eco doppler. El drenaje de estos quistes “de chocolate” puede ser necesaria para prevenir secuelas oculares como pérdida de visión2.
La histocitiosis de células de Langerhans (HCL) también llamada histiocitosis “X” o granuloma eosinófilo, se caracteriza por una proliferación de células de Langerhans, la cual puede ser monofocal o multifocal dependiendo de los sitios de afección. La HCL representa el 1-2% de tumores orbitarios4.
Esta puede ser una enfermedad limitada o monofocal, común en niños entre 2 y 4 años. Regularmente tiene afección ósea, donde se presenta como un aumento de volumen, siendo el cráneo el sitio más afectado. La enfermedad periorbitaria generalmente se presenta dependiendo el sitio de localización de la lesión ocupante, con proptosis en lesiones que ocupan la órbita posterior. Otros signos que podrían aparecer son: dolor localizado en la lesión, fracturas, otitis media, nódulos subcutáneos, ictericia, úlceras en región genital y mucosa oral. La complicación más frecuente es el crecimiento del tumor al interior de la órbita, afectando músculos extrínsecos del ojo e incluso el nervio óptico; por otro lado, existe la posibilidad de infiltración por la fosa orbitaria inferior e irrumpiendo en la fosa craneal media. Esto último podría afectar a la hipófisis, dando como resultado diabetes insípida2.
No se ha podido comprobar la participación de un agente infeccioso en la enfermedad, como ya se había propuesto para CMV, Epstein Barr y VHS-6. Para su diagnóstico son fundamentales estudios de imagen como la TAC y RM, no solo para confirmar la presencia de una lesión ocupante, sino también para determinar la extensión del mismo. Se debe prestar atención a la presencia de erosión en el hueso, así como extensiones intracraneales. La destrucción ósea es relativamente frecuente y se puede apreciar claramente. En este caso, la TAC muestra una masa isodensa moderadamente bien definida, homogénea a los músculos extraoculares. El tratamiento indicado para el rabdomiosarcoma es la extirpación del tumor, ya sea completa o semicompleta. Además de la terapia quirúrgica, la quimioterapia o radioterapia están indicadas, ya que mejoran la tasa de sobrevivencia de los pacientes1. El diagnóstico definitivo se establece mediante biopsia y se basa en la positividad para la proteína S-100 y para el antígeno CD1a o en la visualización de los gránulos de Birbeck en microscopia electrónica. El tratamiento depende de la severidad y extensión sistémica de la enfermedad. Cuando existen lesiones focalizadas, el tratamiento de elección es la biopsia excisional con raspado, con lo que se puede lograr la remisión de la enfermedad. La quimioterapia se utiliza en casos de recidiva o de resecciones incompletas, enfermedad multisistémica o riesgo de afección del sistema nervioso central6,7.
CONCLUSIONES
La histiocitosis de células de Langerhans es una causa infrecuente de tumor orbitario en nuestro medio, teniendo una variedad localizada y multisistémica; esta última involucrando principalmente hígado, bazo y sistema nervioso central teniendo un pobre pronóstico. La forma localizada se caracteriza por lesiones osteolíticas, principalmente en huesos craneales, asociándose a diabetes insípida por infiltración de la hipófisis teniendo un mejor pronóstico. Son fundamentales los estudios de imagen para determinar localización, extensión, estadificación y forma de presentación de la enfermedad. Se considera necesario el seguimiento del paciente para la detección de posibles recidivas. Es de suma importancia estudiar a todos los pacientes con proptosis debido a sus múltiples causas, sugiriendo descartar neoplasias.

