











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink

INTRODUCCIÓN
El oxígeno (O2) es necesario para la vida de la mayoría de los seres vivos, ya que actúa en la respiración mitocondrial como aceptor final de 4 electrones, dando lugar a 2 moléculas de agua y la formación de ATP1,2. Cuando la reducción del O2 es parcial, se generan especies reactivas de oxígeno (ERO) y radicales libres1. De este modo, cuando el O2 capta un electrón, se produce el radical anión superóxido (O2 .-), que puede dar lugar a peróxido de hidrógeno (H2O2) y al radical hidroxilo (OH.)3,4. Otro radical es el óxido nítrico (NO), que al reaccionar con el O2 .- forma especies reactivas de nitrógeno y oxígeno (ERNO) como el anhídrido nitroso (N2O3) y el peroxinitrito (ONOO-)5.
A bajas concentraciones, el NO actúa como un regulador esencial de la presión sanguínea, como protector cardiovascular, como inhibidor de la agregación plaquetaria y también en la adhesión leucocitaria; sin embargo, resulta perjudicial a niveles elevados, ya que afecta el funcionamiento celular mediante la oxidación de proteínas6,7, activa al factor nuclear kappa-B (NF-kB)8 y actúa como mediador de inflamación induciendo a la ciclooxigenasa (COX)9, además de estar involucrado en la apoptosis neuronal, entre otras efectos10.

Es necesario aclarar la diferencia entre una ERO, un radical y un radical libre. Las ERO son compuestos derivados del oxígeno, algunos de los cuales son radicales libres y otros dan origen a alguno ellos11,12. Un radical es una molécula que tiene un electrón desapareado en el último orbital y se define como radical libre cuando se encuentra de manera independiente a otras moléculas. Por ejemplo, el radical hidroxilo (OH.) es una molécula que se deriva del O2, que no está asociado a proteínas y que es capaz de oxidar a los fosfolípidos de las membranas, iniciando reacciones en cadena de lipoperoxidación13.
La producción de ERO se da a nivel subcelular en las mitocondrias, lisosomas, peroxisomas, membrana nuclear y en el citoplasma de diversas células14, en las cuales existen fuentes como las NADPH oxidasas (NOX), una familia de enzimas que utiliza NADPH para reducir al O2, generando O2 .- y la óxido nítrico sintasa (NOS) que produce el NO15,16. De entre todas estas, la fuente de mayor producción de ERO ocurre en la mitocondria, esto debido principalmente, a que cuenta con 11 sitios que pueden generar O2 .- y H2O2. Seis de estos sitios operan con el potencial del par NADH/NAD+ (complejo I) y los 5 restantes operan con el par ubiquinol/ubiquinona (complejo III)17,18(figura 1).
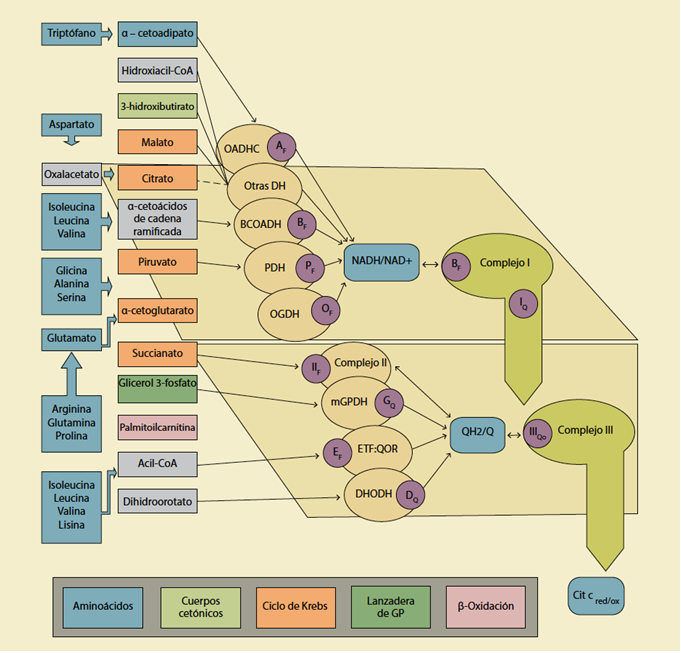
Tomado y modificado de Brand et al., 2015. Se muestran los sitios de producción del O2.- y H2O2 durante el flujo de electrones de la cadena respiratoria a partir de diferentes metabolitos. Los sustratos metabólicos se agrupan en 5 bloques de diferente color, que corresponden al cintillo de la parte inferior de la figura: aminoácidos, cuerpos cetónicos, ciclo de Krebs, lanzadera de glicerol 3 fosfato (GP) y ß-oxidación. En orden descendente se encuentra a las siguientes enzimas: a-cetoadipato deshidrogenasa (OADHC), otras deshidrogenasas (DH), deshidrogenasa de a-cetoácidos de cadena ramificada (BCOADH), piruvato deshidrogenasa (PDH), a-cetoglutarato deshidrogenasa, flavoproteina transportadora de electrones-ubiquinona oxidorreductasa (ETF:QOR), complejo II, glicerol 3 fosfato deshidrogenasa mitocondrial (mGPDH), dihidroorotato deshidrogenasa (DHODH)
Figura 1 Principales sitios de producción de ERO en la cadena respiratoria

EL ESTRÉS OXIDANTE
El estrés oxidante es el desequilibrio entre la generación de ERO y ERNO y los mecanismos antioxidantes en un sistema biológico, donde los primeros sobrepasan la capacidad de las defensas antioxidantes de dicho sistema19. Las ERO y las ERNO eventualmente interactúan con estructuras moleculares, tales como ácido desoxirribonucleico (DNA), proteínas, lípidos y carbohidratos, lo que conduce a alteraciones de la actividad de las vías metabólicas y membranas que, sumado a una inadecuada respuesta antioxidante, causa la acumulación de agregados intracelulares, disfunción mitocondrial, excitotoxicidad y apoptosis. El estrés oxidante se asocia con daño molecular, que eventualmente resulta en algunas patologías como: cáncer, enfermedades neurodegenerativas y diabetes20,21, entre otras (figura 2).
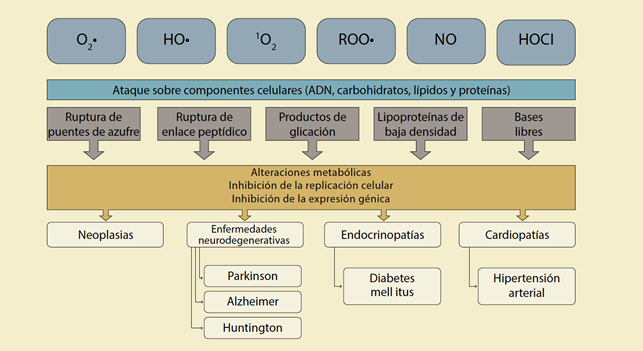
Se especifican algunos de los mecanismos de daño celular asociados al estrés oxidante
Figura 2 El papel de las ERO y ERNO en la fisiopatología de algunas enfermedades
Adicionalmente, el incremento en la producción de radicales libres promueve la entrada masiva de Ca2+ a la mitocondria, lo que hace que se forme el poro de transición en la membrana mitocondrial interna (PTM), llevando a un colapso en el gradiente de potencial electroquímico de protones, lo que provoca una disminución en los niveles de ATP y un aumento en las ERO. La disminución en la producción de ATP resulta en la despolarización de la membrana plasmática y la entrada de Ca+2 a través de varios canales iónicos, lo que produce a la pérdida de la función neuronal y la muerte de la célula22,23.

El estrés oxidante en el sistema nervioso
En el caso del sistema nervioso (SN), las ERO y las ERNO juegan un papel fundamental en el mantenimiento del estado fisiológico, ya que regulan vías de señalización en procesos de supervivencia, desarrollo, plasticidad, muerte e inflamación24,25,26. Sin embargo, el SN es particularmente vulnerable al estrés oxidante debido al elevado consumo de oxígeno que se requiere para mantener su alta tasa metabólica, con la consecuente producción de grandes cantidades de ERO27,28y una menor capacidad de los mecanismos antioxidantes en comparación con otros tejidos29,30.
De esta manera, el estrés oxidante es un factor fundamental en diversos procesos de muerte neuronal, tales como la necrosis y la apoptosis. En la necrosis, la muerte es el resultado de la pérdida de la integridad de las membranas, debido a la lipoperoxidación, el daño oxidante al DNA y a las proteínas estructurales31. Por otro lado, la apoptosis no sólo se caracteriza por la pérdida del potencial de la membrana mitocondrial y la liberación de citocromo C, sino también por la peroxidación proteica que resulta en la disfunción de la ATP sintasa, disfunción de los complejos de la cadena transportadora de electrones y la disminución de la concentración de mecanismos antioxidantes como el glutatión32,33. Las ERO provenientes de las NOX regulan positivamente a la fosfolipasa C y al diacilglicerol, lo que conlleva a un aumento de la concentración intracelular de Ca2+ y a la activación de caspasas efectoras, fenómeno que sin lugar a dudas contribuye significativamente a la muerte celular18 (figura 3).
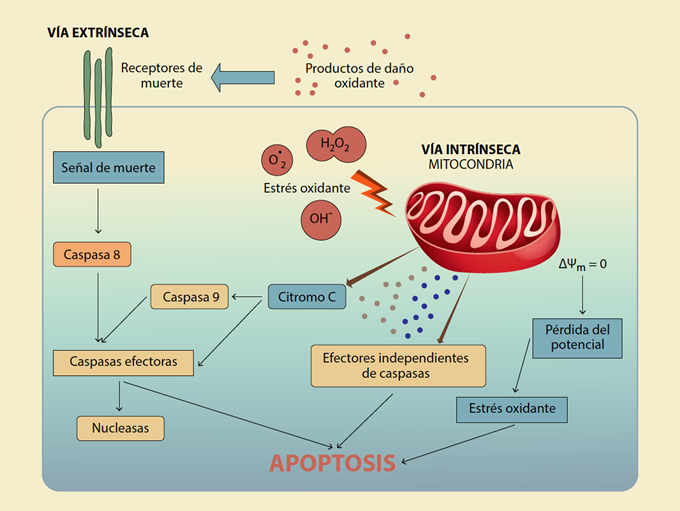
Las ERO participan en las cascadas de señalización que culminan en la muerte apoptótica, ya sea por la producción de daño a biomoléculas, las cuales se liberan al espacio extracelular y son interpretadas como señales extrínsecas de muerte o provocando daño por estrés oxidante, activando vías intrínsecas
Figura 3 Participación de las ERO en el proceso de apoptosis
El daño al SN en las patologías crónicas como las enfermedades de Huntington, Parkinson y Alzheimer, se caracteriza por la muerte neuronal debido al estrés oxidante34, lo que favorece la neuroinflamación que a su vez es responsable de daño secundario. La producción de ERO es un elemento clave en el inicio y la progresión de las enfermedades neurodegenerativas, por lo que resulta susceptible de ser un blanco terapéutico sobresaliente35.
Actualmente se tiene bien dilucidado el papel que tienen los radicales libres y las ERO en las enfermedades neurodegenerativas, y se ha encontrado una especial asociación con las patologías como, la enfermedad de Parkinson (EP), la enfermedad de Alzheimer (EA) y la enfermedad de Huntington (EH), entre otras14.
Según reportes de la Organización Mundial de la Salud (OMS) en el 2006, la enfermedad neurodegenerativa con mayor prevalencia en el mundo es la EA seguida por la EP36, sin embargo, el boletín epidemiológico de México en el año 2015, reporta 2,295 casos de EA, de los cuales, la mayor prevalencia se presenta en pacientes de sexo femenino, mientras que para la EP se reportaron 7,127 casos, con mayor prevalencia en el sexo masculino37,38.
ENFERMEDADES Y ESTRÉS (figura 4)
La enfermedad de Alzheimer
La EA es una enfermedad que se caracteriza por la pérdida gradual de la memoria, que progresivamente compromete otras funciones cognitivas, asociándose a trastornos conductuales que llevan al paciente a un estado de invalidez social y dependencia30,39. Las funciones cognitivas que se alteran en esta enfermedad incluyen la memoria, el lenguaje, la orientación temporo-espacial, las habilidades constructivas, el pensamiento abstracto, la capacidad para resolver problemas y las habilidades motoras adquiridas (praxias), todo esto debido a la muerte de las neuronas piramidales del hipocampo y de la corteza parietal40,41. Entre los hallazgos histológicos propios de la EA se encuentran la acumulación de fibrillas intraneuronales y extracelulares, ovillos neurofibrilares (agregados de proteína tau fosforilada) y placas seniles (agregados filamentosos de péptido beta-amiloide [Aß])42.
La EA está relacionada con una sobreproducción y depósito extracelular de péptido Aß, así como el aumento de la generación de NO, los cuales conducen a vías neurotóxicas que cursan con estrés oxidante, excitotoxicidad e inflamación, que convergen en apoptosis y necrosis por liberación de citocromo C, factor inductor de apoptosis y activación de caspasas43,44.
La proteína precursora de amiloide es una péptido membrana de tejido neuronal y no neuronal (por ejemplo, piel e intestino), que al ser escindida por la β y ϒ secretasa da como resultado el péptido ß amiloide. Este proceso de proteólisis favorece la agregación del péptido ß amiloide en oligómeros solubles que forman fibras que se depositan como placas seniles en el citoplasma de las células neuronales40,45,46,47.
El péptido Aß es capaz de unirse a diferentes componentes mitocondriales, uno de ellos es la proteína de unión β-amiloide alcohol deshidrogenasa (ABAD, por sus siglas en inglés), enzima con papel citoprotector que lleva a cabo la detoxificación de aldehídos como el 4-hidroxi-2-nonenal (4-HNE), un derivado de la peroxidación de lípidos que se utiliza como marcador de estrés oxidante47,48. La interacción entre el péptido Aß y ABAD inhibe a la enzima, lo que conduce a la disfunción mitocondrial y a la generación de ERO48.
El receptor de productos de glicación avanzada se activa por el efecto peroxidante de la proteína Aß, con lo cual se favorece la producción de ERO en la microglía y en las células endoteliales47.
Adicionalmente, el péptido Aß provoca inhibición del complejo IV de la cadena de transporte de electrones y de diversas ATPasas, lo cual contribuye a la generación de ERO y ERNO y a un aumento de la permeabilidad de la membrana mitocondrial interna al Ca2+ contribuyendo a la apertura del PTM47, como parte de una de las vías de señalización proapoptótica en los astrocitos44,49, oligodendrocitos y células endoteliales corticales e hipocampales, lo cual se relaciona con el deterioro de la memoria50,51.
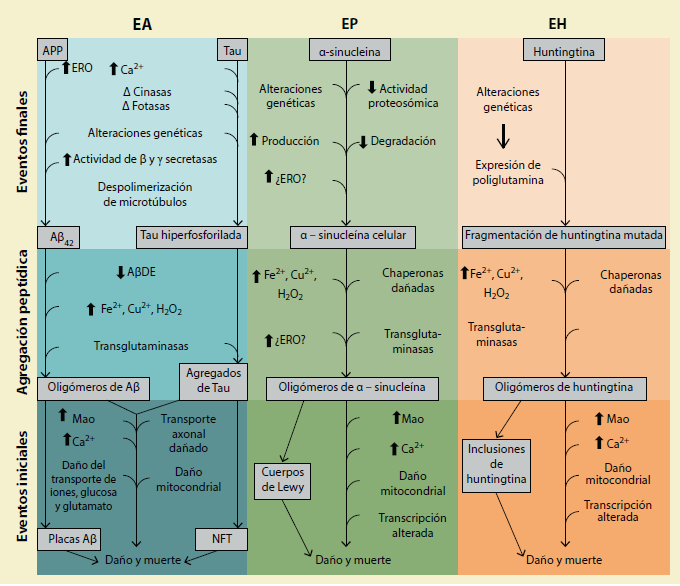
Tomado y modificado de Mark, P. Mattson. Magnus, T. Nature Reviews Neuroscience. 2006. Diferencias entre poblaciones neuronales en la producción y/o eliminación de proteínas anormales que son responsables de la vulnerabilidad neuronal relacionada con la EA, EP y EH. Las proteínas responsables de la fisiopatología en EA son Aβ y tau, en EP la α-sinucleína y en EH la huntingtina. Factores genéticos y relacionados con la edad pueden aumentar las cantidades de proteínas patógenas entre las que se incluyen: Aβ42 en EA-mutaciones en APP o presenilinas (γ-secretasa), especies de oxígeno reactivo (ERO) y reducciones en enzimas que degradan Aβ (AβDE) tales como neprilisina y enzima degradadora de insulina. Tau en EA- ERO, fosforilación y calcio. α-sinucleína en EP-mutaciones en α-sinucleína, parkin, DJ-1, UCH-L1, PINK1 o LRKK-2, ROS y deterioro del proteasoma. EH-expansiones de poliglutamina en huntingtina (Htt), ROS y daño y reparación del ADN. El proceso de agregación de proteínas en sí mismo se ve reforzada por el aumento de la concentración de proteínas, alteraciones postraduccionales tales como modificaciones oxidativas (inducidas por peróxido de hidrógeno, hierro y cobre, por ejemplo) y fosforilación, las acciones del calcio y transglutaminasas y/o proteína chaperona insuficiente. Aunque las proteínas involucradas pueden diferir, existe un considerable sobrelapamiento en los mecanismos por los que dañan y matan a las neuronas
Figura 4 Participación del estrés oxidante en las enfermedades neurodegenerativas (Alzheimer, Parkinson y Huntington)
La enfermedad de Parkinson
La enfermedad de Parkinson (EP) es una de las enfermedades neurodegenerativas con mayor prevalencia en el adulto52. Se caracteriza por degeneración de las neuronas dopaminérgicas ubicadas en la sustancia nigra pars compacta (SNpc). En cortes histológicos post mortem de la corteza cerebral de pacientes diagnosticados con EP se pueden encontrar cuerpos de Lewy (depósitos de α-sinucleína mal plegada, precursor de la neuromelanina, de la cual no se conoce una función específica)53.

La vía anatómica que se encuentra dañada en los pacientes con EP es la vía nigroestriatal, la cual en condiciones normales se encarga del control fino de los ganglios basales54. La disminución en la producción de dopamina en esta vía se manifiesta clínicamente con: temblor en reposo, bradicinesia, rigidez e inestabilidad postural, así como síntomas depresivos y deterioro de la función cognitiva55.
La dopamina es una molécula inestable que se oxida para formar quinonas de dopamina (DAQ, por sus siglas en inglés) y radicales libres, los cuales se incrementan en condiciones de estrés oxidante56,57. El metabolismo de la dopamina que se lleva a cabo por enzimas como la tirosinasa, monoaminoxidasa-B (MAO) y catecol O-metiltransferasa (COMT) en la membrana mitocondrial externa produce H2O2 y ácido dihidroxifenilacetaldehído56,58,59. Si las barreras antioxidantes glutatión (GSH), glutatión peroxidasa y la glutatión peroxidasa no son capaces de reducir el H2O2, este puede reaccionar con metales de transición como el hierro para formar radicales OH., desencadenado la peroxidación lipídica de la membrana y la subsecuente muerte celular60,61.
Las ERO y DAQ pueden modificar grupos sulfhidrilo en las proteínas de la cadena transportadora de electrones y afectar su actividad62,63. Además, la modificación de grupos sulfhidrilo puede inducir la apertura del PTM. Esto se ha caracterizado ya que se puede prevenir la apertura del PTM64 y la muerte neuronal al administrar NAD(P)H quinona reductasa (NQ01) in vivo a pacientes65,66.
Por otro lado, las DAQ puede ciclarse, y formar aminocromos, que son altamente reactivos, por lo que conducen a la generación de O2 . y agotamiento del NADPH54; Finalmente, los aminocromos puede formar aductos con proteínas tales como la α-sinucleína, que se relaciona con el estrés oxidante por un aumento de NO, O2 ∙ y H2O266,67.

Enfermedad de Huntington
La enfermedad de Huntington (EH) se debe a la repetición del triplete: citosina, adenina y guanina (CAG) en el gen HD, localizado en el brazo corto del cromosoma 4, que codifica para una proteína rica en residuos de glutamina, llamada huntingtina (HTT). Por lo anterior esta se considera una enfermedad hereditaria, con patrón autosómico dominante68,69. En condiciones fisiológicas, el número de repeticiones del triplete CAG oscila entre 9 y 36, mientras que la forma mutada de la proteína huntingtina (mHTT) tiene más de 36 repeticiones, lo que favorece su agregación y toxicidad en las células del sistema nervioso, principalmente las neuronas GABAérgicas del núcleo estriado (caudado y putamen)70,71,72.
La función de la proteína HTT no se ha aclarado completamente, aunque se propone que participa en el movimiento de los microtúbulos y las vesículas sinápticas que contienen al factor neurotrófico derivado del cerebro (BDNF)73.
La enfermedad se caracteriza por deterioro motor, cognitivo y cambios en la personalidad. Se manifiesta por movimientos involuntarios (corea), que son cada vez más frecuentes, involucrando grupos musculares de cabeza, cuello y extremidades, lo que afecta sus actividades cotidianas, llevándolo a la pérdida de la autonomía y posteriormente a la muerte74.
Las manifestaciones de la enfermedad pueden comenzar en cualquier momento de la vida; sin embargo, se presenta con mayor frecuencia entre los 35 y 50 años de edad. Los pacientes con EH cursan con neurodegeneración progresiva que los conduce a la muerte entre 15 y 20 años después de su diagnóstico. Actualmente el tratamiento se ve limitado al control de los síntomas75,76.
El número de repeticiones de CAG en el gen HTT es el principal factor de predicción de la edad de inicio de la enfermedad. Los pacientes con penetrancia reducida (entre 36 y 39 repeticiones) pueden o no presentar la enfermedad77,78. Cuando la penetrancia es completa (más de 39 repeticiones) los pacientes presentan los síntomas de la enfermedad entre los 35 y 50 años79,80, es importante mencionar que los pacientes con inicio temprano de la enfermedad presentan más de 60 repeticiones81.
Algunos cambios asociados a la EH son el aumento de lactato secundario a la disminución del catabolismo aerobio de la glucosa, particularmente en regiones como la corteza cerebral y los ganglios basales; también se han encontrado cambios morfológicos en las mitocondrias de neuronas corticales, disminución de la fosforilación oxidativa por reducción de la actividad de la cadena transportadora de electrones (complejos II, III, y IV) y despolarización de la membrana mitocondrial81.
En cultivos celulares el aumento en la agregación de mHTT produce cambios mitocondriales, como la reducción de la capacidad de la homeostasis del Ca2+ ligado a la excitotoxicidad mediada por los receptores tipo NMDA52,82. Los cambios mitocondriales y la subsecuente mitofagia asociada con la agregación de mHTT son los responsables de la producción de ERO83.
El daño oxidante se ha documentado en el tejido cerebral post mortem, linfoblastos y líquido cefalorraquídeo. Algunos de los indicadores de daño oxidante que se han observado en el cuerpo estriado y corteza cerebral de estos pacientes son el aumento en la concentración de malondialdehído (MDA) y 4-hidroxinonenal (productos de oxidación lipídica), incremento en la carbonilación y la nitración de las proteínas83,84, así como la disminución en el glutatión reducido e incremento de la glutatión peroxidasa, catalasa y superóxido dismutasa85.

EL PAPEL DE LOS ANTIOXIDANTES EN LAS ENFERMEDADES NEURODEGENERATIVAS
Al tomar en cuenta el papel de EROS y ERNO en las enfermedades neurodegenerativas la manipulación de los niveles de los mismos parece ser un tratamiento prometedor para frenar la neurodegeneración41. Se propone que los antioxidantes pueden:
Disminuir la concentración de oxidantes.
Unirse a iones metálicos para evitar la formación de especies reactivas.
Transformar los peróxidos en productos menos reactivos.
Detener la propagación y el aumento de radicales libres.
Los antioxidantes se han clasificado en 2 principales sistemas: el enzimático (superóxido dismutasa, glutatión peroxidasa y catalasa, entre otros) y no enzimático (vitamina E, vitamina C, glutatión, ácido lipoico, carotenoides y ubiquinona, entre otros)86,87 (tabla 1).
Tabla 1 Antioxidantes no enzimáticos y sus mecanismos propuestos en el tratamiento de EH, EA y EP
| Antioxidante | Mecanismo de acción | Efecto terapéutico propuesto | Cita |
|---|---|---|---|
| Vitamina E | Evita lipoperoxidación | Reduce el riesgo de EA y EP | 85 |
| Vitamina C | Eliminación del anión superóxido mediante la formación de semidehidroascorbato, el que es reducido por el glutatión | En uso conjunto con la vitamina E disminuye el depósito del péptido Aß en la EA, y favorece el retraso en la pérdida neuronal en EA y EP | 86,87 |
| Glutatión | Destoxificante de radicales libres, peróxidos y xenobióticos | Disminuye la pérdida neuronal en la EA | 27, 86 |
| Ácido lipoico | Coenzima mitocondrial con acciones antioxidantes y quelante de metales | Retrasa la pérdida neuronal en la EA y la EP | 87 |
| Carotenoides | Evitan la lipoperoxidación | En conjunto con la vitamina E previene la pérdida neuronal, así como la peroxidación lipídica en mesencéfalo y cuerpo estriado en la EP | 85, 27 |
| Ubiquinona (CoQ10) | Inhibición de la lipoperoxidación | En uso conjunto con la vitamina E y la C retrasa el inicio de la disminución cognitiva en EA, EH y EP | 21, 84, 85 |
EA: enfermedad de Alzheimer; EH: enfermedad de Huntington; EP: enfermedad de Parkinson.
CONCLUSIONES
El inicio del estrés oxidante es clínicamente imperceptible, es decir, no existe un estudio de laboratorio clínico con el que se pueda determinar la concentración de radicales libres, ERO o ERNO en el organismo, por lo cual se dificulta el actuar específicamente a este nivel en las enfermedades neurodegenerativas. Aunque se ha propuesto la suplementación con antioxidantes para contrarrestar la producción de radicales libres, los resultados de estos estudios son controversiales. Por lo anterior, es importante dilucidar el papel de los radicales libres en los procesos neurodegenerativos con el fin de tener indicios sólidos sobre las posibles dianas de tratamiento con la intención de retrasar o prevenir la progresión del daño en este tipo de enfermedades.

