











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La neuromielitis óptica (NMO) o enfermedad de Devic, es un padecimiento infrecuente de carácter inflamatorio del sistema nervioso central (SNC) que afecta principalmente al nervio óptico y a la médula espinal, aunque también existen estructuras cercanas al sistema ventricular involucradas. Actualmente se concibe como una enfermedad independiente de la esclerosis múltiple (EM), aunque por muchos años se consideró como una de sus variantes clínicas1. El mecanismo de la enfermedad está dado por lintocitos B que producen anticuerpos IgG dirigidos contra el canal de acuaporina 4 (AQP4), que está presente en abundancia en las prolongaciones pediculares de los astrocitos en la barrera hematoencefálica2,3. Dichos anticuerpos desencadenan un proceso inflamatorio y consecutivamente desmielinizante localizado en el nervio óptico, médula espinal y las estructuras circunventriculares, sobre todo aquellas cercanas al piso del cuarto ventrículo, como el área postrema, entre otras4.
La presentación clínica consiste en neuritis óptica o mielitis, cuya recuperación suele ser escasa o nula. Las técnicas de imagen muestran frecuentemente lesiones medulares longitudinales y extensas que abarcan 3 o más segmentos vertebrales. Es de vital importancia el diagnóstico diferencial inicial con EM y otros trastornos autoinmunes dado que el tratamiento de muchos de estos padecimientos pueden agravar la NMO5.
CASO CLÍNICO
Se presenta el caso de una mujer de 55 años de edad, sin antecedentes de importancia, quien inició a los 41 años con parestesias en ambos miembros torácicos de forma intermitente. Después de unos meses, dichos síntomas progresaron a alodinia fluctuante sin mejoría con el uso de analgésicos. Dos años después presentó un nuevo evento manifestado por paraparesia e hipoestesia de tronco y miembros pélvicos, el cual limitaba la deambulación; además de la disminución progresiva de la agudeza visual, que motiva acudir a una nueva valoración médica y fue enviada días después al departamento de neurología.
En la evaluación inicial a la exploración no se observaron anormalidades. Neurológicamente, la prueba de Mini-mental tuvo un resultado de 30 puntos, con amaurosis del ojo derecho, distinguía solo luces y sombras en ojo izquierdo, y presentaba un defecto pupilar aferente y papilitis en ambos ojos.
A nivel motor, presentaba espasticidad leve de miembros pélvicos Ashworth 2, con fuerza 4/5 de miembros torácicos y 2/5 de miembros pélvicos, además de hiperreflexia generalizada, con signo de Babinski, Hoffman y Trömner bilaterales. Al evaluar la sensibilidad, se encuentra hipoestesia de extremidades superiores a partir de C5 así como en extremidades pélvicas, hipopalestesia en las 4 extremidades y pérdida de los reflejos abdominocutáneos.
Como parte del abordaje diagnóstico se realizaron estudios de laboratorio completos, medición de inmunoglobinas, panel viral, panel inmunológico (anticuerpos anti-ácido desoxirribonucléico [anti-DNA] de doble cadena, antipéptidos cíclicos citrulinados [anti-CCP], anti ribonucleoproteína [anti RNP], anti Ro, células de lupus eritematoso [LE], entre otros), que se reportaron como normales o negativos, según fuera el caso. Se practicó una punción lumbar que mostró líquido cefalorraquídeo (LCR) normal con ausencia de bandas oligoclonales. Se realizó un estudio de resonancia magnética de columna cervical y encéfalo donde se observó (en secuencia T2) una lesión hiperintensa intramedular cervical menor a un segmento vertebral de extensión y otra lesión hiperintensa intramedular torácica extensa de más de 3 segmentos vertebrales de extensión (Figura 1), en encéfalo (en secuencia FLAIR) se hizo evidente el hallazgo de lesiones hiperintensas periventriculares confluentes y escasas de sustancia blanca sin edema perilesional (Figura 2).
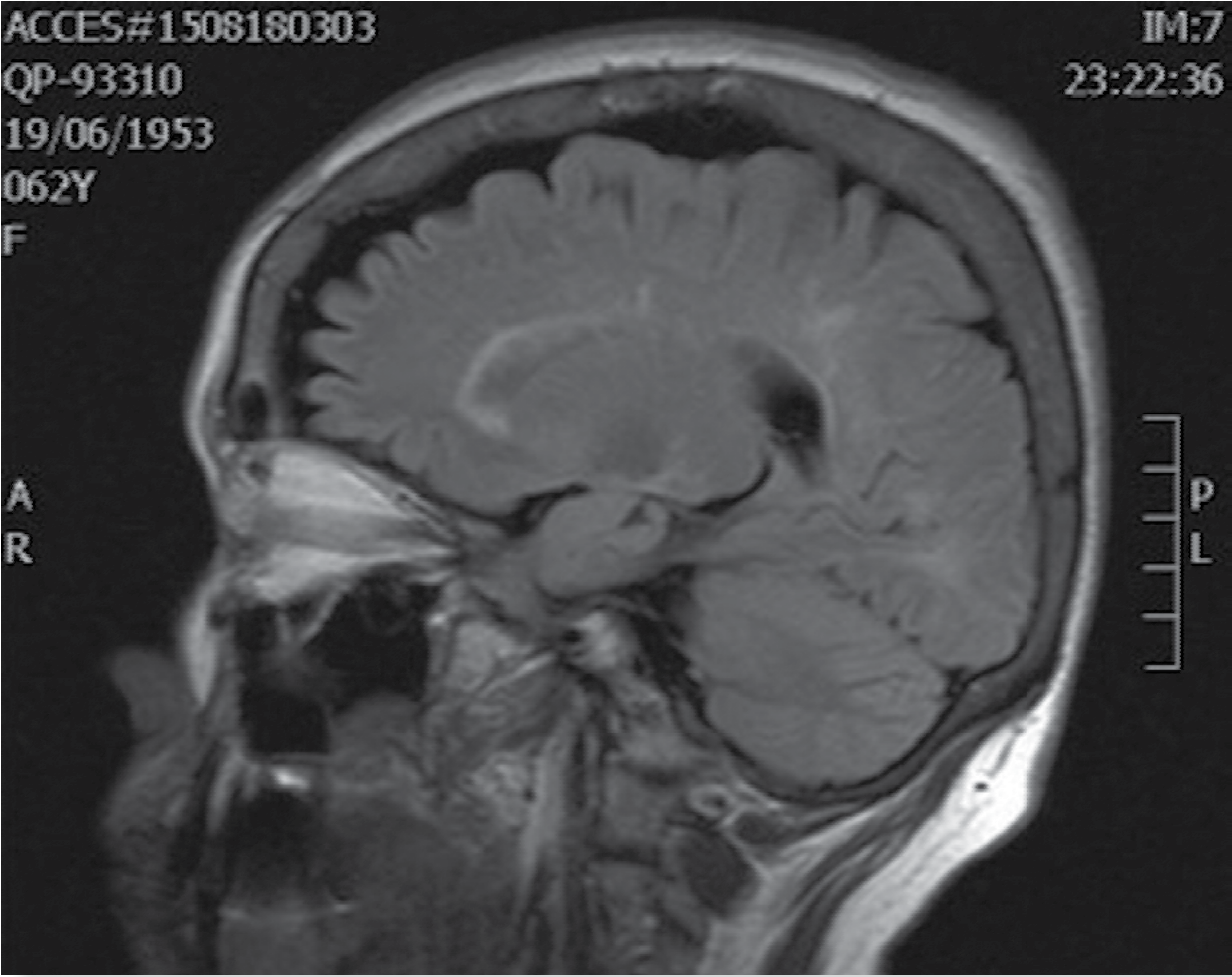
Foto: otorgada por los autores
Figura 1 Imagen de resonancia magnética nuclear de encéfalo en secuencia FLAIR que evidencia zonas hiperintensas de desmielinización periventricular.

Foto: otorgada por los autores
Figura 2 Imagen de resonancia magnética nuclear de médula espinal cervical y torácica alta en corte sagital en secuencia T2 en la que se aprecia una pequeña lesión hiperintensa cervical posterior y otra longitudinalmente extensa abracando más de 3 segmentos vertebrales y se continúa a lo largo de la médula dorsal.
Se inició manejo con inmunoglobulina intravenosa en busca de un efecto inmunomodulador, con mínima mejoría de los síntomas sensitivos y, dado las características clínicas y hallazgos en la neuroimagen, se realizaron anticuerpos Anti-NMO con resultado cualitativo positivo, obtenido semanas después, aunque la sospecha clínica era evidente. Por lo anterior, se concluyó el diagnóstico de NMO y se continuó el tratamiento con anticuerpos monoclonales (rituximab). La paciente presentó mejoría parcial en la sintomatología, principalmente en la sensibilidad y mínimo en la fuerza muscular. Después de un año de seguimiento y con un esquema ya establecido de rituximab ha continuado sin nuevas recaídas, aunque con discapacidad importante debido a las secuelas, sin progresión de la enfermedad y con estudios de neuroimagen sin cambios y sin evidencia de actividad de la enfermedad (lesiones nuevas, captación de contraste o extensión de las lesiones previas).
ANÁLISIS Y REVISIÓN DE LA LITERATURA
Epidemiología
La NMO es una enfermedad con creciente interés en el campo de la neurología. Existen pocos estudios acerca de su verdadera prevalencia e incidencia a nivel mundial. Hay reportes que estiman una prevalencia de entre 0.51 a 4.4 casos por cada 100,000 habitantes, y es mayor en las mujeres6,7. A diferencia de la EM, el debut de la enfermedad se da a mayor edad, con una edad media de inicio entre los 30 y 40 años, donde se dan los ataques más graves y con menor probabilidad de recuperación8,9. Los pacientes mayores comúnmente se presentan con mielitis, mientras que los mas jóvenes debutan con neuritis óptica10.
Es más susceptible la población no blanca, que incluye aquellos descendientes africanos (afrocaribeños y afroamericanos), hispanos, asiáticos y americanos nativos 6,11.
En México no existen estudios que determinen la prevalencia o incidencia exacta de esta enfermedad. Un estudio realizado por médicos del Instituto Nacional de Neurología y Neurocirugía “Manuel Velasco Suárez” estima que la incidencia aproximada es de 1.3 casos por cada 100,000 habitantes. Sin embargo, debemos considerar que la mayoría de los datos son recopilados de series de casos y no de estudios epidemiológicos12.
Fisiopatología
El fenómeno inicial para la producción de anticuerpos AQP4 por los linfocitos B es desconocido con evidencia de la participación de interleucina17, y con la sospecha de una producción predominantemente en la periferia dada su mayor concentración en suero4,6,7,9. Estos anticuerpos pueden entrar al SNC debido a una deficiente barrera hematoencefálica o por una ruptura de ésta, dando lugar al reconocimiento del antígeno (AQP4) localizado de forma abundante en el nervio óptico, la médula espinal, estructuras periventriculares (vecinas al tercer y cuarto ventrículo) hipotálamo, tálamo y cerebelo, entre otras.
La AQP4 es una proteína canal de agua en las prolongaciones pediculares de los astrocitos, nervios óptico y superficie pial y ependimal en contacto con el LCR, aunque también está presente fuera del SNC, como en las células epiteliales del riñón, el estomago, vías respiratorias, glándulas y musculo esquelético13. Una vez que existe esta reacción antígeno-anticuerpo ocurre un proceso inflamatorio que incluye la activación del sistema del complemento junto con la migración de macrófagos, neutrófilos y eosinofilos con rompimiento de la barrera hematoencefalica debido a una cascada inflamatoria local, dando lugar a una desmielinización perivascular que se acompaña de pérdida axonal, necrosis, y edema intramielina, observándose en lesiones crónicas cavitaciones y atrofia tisular importante4,9, datos que suelen ser infrecuentes en la EM, en donde no se ha encontrado un antígeno especifico y con la particularidad de considerarse una enfermedad con predominio de la inmunidad de tipo celular por linfocitos T CD43,4.
Clínica
La NMO y los trastornos del espectro de la NMO pueden presentar diferentes variantes clínicas, dentro de las que se incluyen formas limitadas (con neuritis o mielitis aislada), síndromes de afección del tallo cerebral o área postrema, síndromes diencefálicos agudos, mielitis longitudinalmente extensa asociada a enfermedades autoinmunes y neuritis óptica o mielitis asociada a lesiones de NMO14,15.
Los pacientes suelen tener una edad discretamente mayor que los de EM y presentan síntomas que de forma predominante suelen ser agudos o subagudos con evolución progresiva, caracterizándose por pérdida de la visión (neuritis óptica), bilateral (a diferencia de la EM, que suele ser unilateral y con tendencia a la remisión con o sin tratamiento) y no dolorosa, o mielitis que se traduce en tetraparesia o paraparesia con manifestaciones sensitivas y motoras e incluso con alteración del control de esfínteres, a diferencia de la EM que suelen presentar síntomas de predominio sensitivos debido a una mielitis transversa incompleta16. Los síntomas como el vómito intratable, náusea y tos persistente son los más frecuentes en la afección encefálica17.
Diagnóstico
Los primeros criterios diagnósticos fueron elaborados en 1999, posteriormente en el 2006 se complementaron y después de distintas modificaciones en el año 2015 se afinaron diferentes puntos que incluyen ahora a los pacientes con presentaciones clínicas (nausea, vómito o tos incoercible, síncopes de repetición, entre otros) que previamente no se consideran dentro del diagnóstico de NMO, pero que hasta el día de hoy forman parte de los ya conocidos trastornos del espectro de la NMO (Tabla 1)15.
Tabla 1 Criterios actuales del consenso de 2015 para el diagnóstico de trastornos del espectro de neuromielitis óptica, publicados por Wingechuck M et al.14
AQP4: aquaporina 4; IgG: inmunoglobulina G; MTLE: mielitis transversa longitudinalmente extensa.
Después de los hallazgos clínicos que son diferentes en comportamiento y presentación con la EM, lo estudios de extensión con respecto al laboratorio, en suero, suelen ser normales, excepto por la presencia del anticuerpos contra AQP4, punto importante en los criterios diagnósticos de NMO15, de la misma forma el estudio de LCR que puede ser normal o presentar datos inespecíficos de inflamación manifestados por pleocitosis y elevación de proteínas, con menor frecuencia la presencia de bandas oligoclonales (presentes en un 15%) a diferencia de la EM, en donde estas últimas suelen presentarse hasta en el 85% de los pacientes.
Con respecto a los estudios de imagen la tomografía axial de cráneo no es útil como apoyo diagnóstico, pero la resonancia magnética de encéfalo y médula espinal es vital para el diagnóstico. Al inicio el estudio puede ser normal, pero cuando existen alteraciones (más evidentes en las secuencias T2 y FLAIR), un criterio es que no sean compatibles con lesiones características de la EM, sucediendo en mayor frecuencia lesiones medulares centrales extensas (> 3 segmentos vertebrales, inusuales en la EM) que pueden esparcirse incluso hasta el tallo, y en el caso de episodios de neuritis óptica aguda es posible observar captación del medio de contraste (gadolinio) en el nervio óptico. Otras lesiones que pueden encontrarse son hiperintensidades cercanas al tercer o cuarto ventrículos (tallo cerebral) y lesiones hipotalámicas, talámicas, cerebelosas o inespecíficas de sustancia blanca1,3,14,18.
Tratamiento
Debido a que no existen estudios aleatorizados doble ciego que ayuden a guiar la pauta terapéutica en estos pacientes, la mayoría de las sugerencias en el tratamiento se ha obtenido de estudios observacionales, descriptivos y series de casos.
El tratamiento incluye 2 etapas: el manejo del ataque agudo y la terapia de mantenimiento. Todos los ataques deben recibir tratamiento inmediato utilizando dosis altas de esteroides, extrapolándose a las recaídas de la EM con metilprednisolona a dosis de 1 g diario por 5 días con el fin de suprimir la respuesta inflamatoria; en situaciones alternas se puede utilizar plasmaféresis, dada su efectividad comprobada4.
La inmunoglobulina puede también ser útil en el manejo de recaídas; sin embargo, la evidencia reportada hasta ahora es escasa19. Con respecto al tratamiento de mantenimiento inmunomodulador, se ha propuesto la prednisona (1 mg/kg) debido a su rápido efecto y especialmente después de haber confirmado el diagnóstico con reciente evidencia de actividad de la enfermedad.
Convendrá después continuar con un inmunosupresor ahorrador de esteroide, con el objetivo de disminuir los efectos adversos asociados al uso crónico de esteroides, por lo que se utiliza comúnmente azatioprina o micofenolato, los cuales tienen eficacia completa hasta 6 meses después de haberse iniciado, pero cabe mencionar que predictivamente los pacientes con historia de recaídas severas e inicio de la enfermedad en edad joven tienen un alto riesgo de pobre respuesta a estos 2 tratamientos20. Por otro lado, el rituximab, un anticuerpo monoclonal anti-CD20 es otra alternativa de primera línea de tratamiento que ha tenido muy buen perfil de seguridad y eficacia en diferentes esquemas descritos, aunque con mayor frecuencia se indica en aplicaciones semestrales; no obstante, su principal desventaja es el costo4.
En general, el pronóstico no es muy favorecedor, más aún cuando el tratamiento se ha iniciado después de haber existido frecuente actividad de la enfermedad, pues los pacientes suelen permanecer con muchas secuelas, incluso a pesar de recibir tratamiento durante las recaídas de la enfermedad21.
Conclusiones
La NMO es una enfermedad con prevalencia e incidencia escasamente determinadas, cuya presentación clínica representa un reto para su sospecha y diagnóstico diferencial; sin embargo, en el momento en que un paciente cumple con algunas de las características para sospecha de NMO (sexo, edad, presentación clínica, respuesta parcial o pobre a tratamiento en recaídas, entre otras) debe derivarse de inmediato a un servicio de neurología, ya que se ha reportado un retraso promedio de un año en el diagnóstico en la mitad de pacientes con NMO confirmada8, lo que da lugar a una menor tasa de recuperación y un retraso en el inicio del tratamiento.
Similar a la historia del caso que se presenta, existe un subdiagnóstico de este padecimiento o bien se cataloga como EM y consecutivamente se trata como tal, por lo que es fundamental reconocer las características clínicas, de imagen y laboratorio para agilizar la detección y el inicio de tratamiento inmunomodulador de forma oportuna con el objetivo de evitar secuelas debido a la continua actividad y severidad de la enfermedad a largo plazo.

