










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Facultad de Medicina (México)
On-line version ISSN 2448-4865Print version ISSN 0026-1742
Rev. Fac. Med. (Méx.) vol.55 n.2 Ciudad de México Mar./Apr. 2012
Artículo de revisión
Leucemia para el médico general
Leukemia for the general practitioner
Rafael Hurtado Monroya, Braulio Solano Estradab, Pablo Vargas Viverosc
a Jefe del Servicio de Hematología. Hospital Ángeles del Pedregal. México, DF. Correo electrónico: rafahurtado@prodigy.net.mx
b Medicina Interna. Hospital Ángeles del Pedregal. México. DF.
c Medicina Interna. Hospital Ángeles del Pedregal. México. DF.
Recibido: 17 de octubre 2011
Aceptado: 07 enero 2012
INTRODUCCIÓN
A pesar de los grandes avances moleculares y terapéuticos en el estudio de las leucemias, los aspectos básicos de este padecimiento aún no se conocen de manera clara por el médico no hematólogo, por lo que el objetivo de este trabajo es proporcionar información fundamental a los estudiantes de medicina y médicos en general, y que permita sobre todo obtener el conocimiento general de las leucemias, su diagnóstico oportuno y buscar la referencia temprana con el hematólogo.

DEFINICIÓN
Leucemia es el término que se utiliza para definir a un grupo de enfermedades malignas de la sangre. El diagnóstico temprano es esencial, ya que le permitirá al paciente acudir de manera temprana con el médico especialista en hematología, quien conducirá el proceso diagnóstico y ofrecerá el tratamiento específico. Se caracteriza por tener una proliferación clonal, autónoma y anormal de las células que dan origen al resto de las células normales de la sangre (comportamiento tumoral en general).
Lo anterior implica que una célula temprana sufre un cambio genético que hará que se produzca sin control una clona (colonia) anormal de sí misma. Esta producción anormal es desordenada porque las células anormales se multiplican en imagen y semejanza de ellas mismas, por lo que ocupan paulatinamente el espacio de la medula ósea normal y provocan anemia progresiva, sangrado anormal y predisposición a las infecciones. Por otro lado, cuando las células anormales invaden otros tejidos, se producirá falla del funcionamiento del órgano que se ocupa, por ejemplo, la infiltración al sistema nervioso central que ocurre en la leucemia aguda linfoblástica (LAL) se podría manifestar con cefalea, crisis convulsivas, alteraciones motoras focalizadas, aumento de la presión intracraneana, y de no hacer el diagnóstico temprano y proporcionar el tratamiento adecuado, presentará pérdida de la función y consecuencias irreversibles.
MANIFESTACIONES CLÍNICAS
El cuadro clínico es diverso y dependerá del tipo de leucemia: aguda o crónica, sin embargo para las 2 existen manifestaciones clínicas inespecíficas (que ocurren en cualquier enfermedad):
1. Fatiga.
2. Cansancio fácil.
3. Debilidad generalizada.
4. Deseos de permanecer en reposo o en cama.
5. Requiere de la ayuda de alguien para satisfacer sus necesidades personales.
Las leucemias crónicas son de curso indolente y hasta un 50% de los casos se descubren en una revisión clínica de rutina o de laboratorio en voluntarios que se consideran sanos y acuden a donar sangre, sin embargo, conforme progresa la enfermedad, se presentan las manifestaciones inespecíficas pero ahora son específicas (tabla 1).

En las formas agudas, las manifestaciones específicas se derivan de la deficiencia de alguna de las líneas celulares:
1. Eritrocitos: síndrome anémico cuya intensidad dependerá del grado de hipoxemia sin importar el grado de anemia. Disnea de medianos esfuerzos hasta la ortoprea.
2. Plaquetas: petequias, equimosis en extremidades, y en casos más graves generalizados, hemorragia seca y húmeda con epistaxis, gingivorragia, hematuria, melena o hematoquesia. Muy grave en el sistema nervioso central (SNC).
3. Leucocitos: fiebre, diaforesis, infecciones localizadas hasta una franca septicemia (bacterias u hongos). Ocurren con neutropenia menor a 250 neutrófilos/mm3 totales.
Síndrome infiltrativo: se refiere a la implantación anómala en cualquier tejido, aunque lo frecuente es:
1. Hepatomegalia o esplenomegalia (figura 5).
2. Adenomegalia (local o generalizada).
3. Cutis leucémica.
4. Dolor óseo por expansión de la médula ósea.
5. Tejidos blandos (sarcoma granulocítico).
6. Testicular.
7. SNC.
8. Encías y cualquier sitio (figura 1).

Trastornos metabólicos: resultan de la hiperproducción anormal de células malignas y el aumento de apoptosis.
1. Acidosis.
2. Aumento de la deshidrogenasa láctica (DHL).
3. Hiperkalemia.
4. Hiperuricemia.
5. Aumento de la β2 microglobulina.

La evidencia clínica predomina como la piedra angular de la sospecha diagnostica de las leucemias y de cualquier padecimiento, pero lo que sigue es complementar el diagnóstico con el apoyo del laboratorio clínico en la citometría hemática completa o especial, lo que quiere decir, la observación minuciosa del frotis de sangre periférica por personal técnico que tenga la preparación en la identificación de células anormales y sobre todo leucémicas.

Las alteraciones del laboratorio que obligan a una revisión especial incluyen:
1. Anemia (cualquier grado).
2. Leucopenia o leucocitosis (predominio de una línea celular).
3. Trombocitopenia.
4. Combinaciones: bicitopenia o pancitopenia.
Se debe tener especial cuidado cuando el laboratorio reporta la presencia de leucocitos o linfocitos atípicos (pueden ser blastos leucémicos). Es recomendable solicitar la revisión de un experto (figura 2).

El aspirado de médula ósea es indispensable para en el diagnóstico (figura 3) y se requiere de un 20% de blastos para establecer el criterio de leucemia aguda en cualquiera de sus variedades.

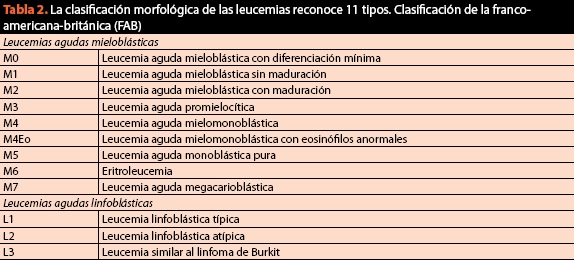
En el mismo procedimiento se deberán obtener muestras para la clasificación final del padecimiento y solicitar cariotipo e inmunofenotipo, ya que actualmente el criterio citomorfológico es de vital importancia pero ya no es suficiente. En la (tabla 2) se enlista la clasificación vigente y actual de los padecimientos malignos de la sangre.
El tratamiento está dirigido hacia 2 aspectos importantes: el primero de ellos es el específico antileucémico y se basa en el uso de medicamentos de origen químico que se les conoce con el nombre de quimioterapia, cuyo principal objetivo es erradicar, es decir, eliminar a todas las células leucémicas del organismo. El segundo aspecto del tratamiento es el apoyo para las complicaciones que por lo general presentan los pacientes en su ingreso.
El tratamiento está dirigido hacia 2 aspectos importantes: el primero de ellos es el específico antileucémico y se basa en el uso de medicamentos de origen químico que se les conoce con el nombre de quimioterapia, cuyo principal objetivo es erradicar, es decir, eliminar a todas las células leucémicas del organismo.
El segundo aspecto del tratamiento es el apoyo para las complicaciones que por lo general presentan los pacientes en su ingreso como son:
1. Anemia.
2. Hemorragia anormal.
3. Infecciones pulmonares y generalizadas, entre otras (figura 4).
4. Cualquier otra complicación adyacente que el paciente pueda tener (co-morbilidad), como padecimientos preexistentes, por ejemplo, diabetes, hipertensión, cardiopatías y otras enfermedades frecuentes entre los pacientes que sufren de leucemia.


Por lo anterior, es muy importante tener en cuenta que el tratamiento en contra de la leucemia es multidisciplinario, que implica la participación de otros especialistas como apoyo al hematólogo.
El tratamiento antileucémico también será diferente para los distintos tipos de leucemia y para las formas agudas. Se divide en 3 fases:
1. Inducción de la remisión. El objetivo es llegar a la remisión completa (RC), es decir, la normalización de los valores de la sangre del paciente, la ausencia de cualquier síntoma o signo de que la leucemia persista con infiltración. Durante el proceso el paciente deberá tener un "estado libre de leucemia" en la Médula ósea y el futuro deberá ser la recuperación a una hematopoyesis normal, y desafortunadamente en otros casos, se recuperan con la enfermedad, lo cual habla de leucemia resistente cuyo pronóstico es pésimo. Este primer proceso puede llevar de 6 a 8 semanas para lograr la RC.
2. Consolidación. Implica el uso de los mismos medicamentos que se usaron en la inducción o la combinación de otros quimioterápicos, también con el propósito de seguir la erradicación de las células malignas residuales que pudieran desarrollar resistencia a los de primer uso.
3. Mantenimiento. Se prefiere mantener al paciente bajo el efecto de quimioterapia ante la posibilidad de actividad leucémica incipiente y que con el tratamiento mantenga efecto hasta desaparecer la enfermedad.

Hasta el momento para las formas agudas, los criterios más estrictos para considerar RC se vuelven más complejos, ya que lo más recomendable es buscar la remisión molecular, que implica la búsqueda de la alteración cromosómica inicial como ocurre en el caso de la leucemia promielocítica (M3-FAB) con t(15;17) inicial, se deberá buscar por estudios moleculares y citogenéticos específicos a pesar de estar en RC, ya que de persistir la traslocación, entonces se deberá continuar el tratamiento agresivo hasta la eliminación total de la clona. Se debe considerar a esta variedad de leucemia como potencialmente curable y la cual es, de la más frecuente en la población latina.
La curación del padecimiento dependerá entonces de la eliminación de todas las células malignas existentes en el paciente. En general, algunas de las leucemias pueden ser susceptibles a la curación con la quimioterapia solamente, pero hoy día se debe dar mucha importancia a los llamados factores pronósticos que se basan en modelos matemáticos que permiten ubicar a los pacientes en el grado de pronóstico que tienen e incluyen:
1. El tipo de leucemia.
2. La alteración molecular inicial y su persistencia a pesar del tratamiento o su erradicación.
3. La Edad. Los pacientes mayores de 60 años tienen un pronóstico malo en comparación con los pacientes de menor edad.
4. La quimioterapia. Se deberán utilizar los medicamentos indicados, y sobre todo, las dosis recomendadas. Por ejemplo en la LAL del adulto, el utilizar el esquema HyperCyVAD (dosis escaladas de ciclofosfamida, vincristina, adriamicina, dexametasona en combinación con arabinosido de citosina y methotrexate) logra un 90% de RC y cura en el 50% de los casos, datos no antes vistos con otros esquemas. Este tratamiento es tóxico y requiere que su uso sea sólo en instituciones que tengan los recursos de apoyo suficientes. Desafortunadamente, en nuestro medio no todos los centros cuentan con los medicamentos recomendados y los resultados no serán reproducibles, ya que tampoco cuentan con suficiente apoyo para el paciente durante la fase de máxima mielosupresión.
5. Terapia de apoyo. Los logros de la quimioterapia y los nuevos medicamentos con nuevas combinaciones y con más especificidad obligan la implementación de los grupos multidisciplinarios con la dirección del hematólogo; instalación y uso de catéteres centrales; apoyo de banco de sangre para el soporte de transfusiones de plaquetas y eritrocitos (incluso productos radiados); la intervención del infectólogo para la detección de infecciones, y el uso adecuado de antibióticos o antimicóticos, ya sea profilácticos o terapéuticos, en caso necesario; la habilitación de cuartos de aislamiento con servicios de mantenimiento e intendencia que permitan lograr un ambiente libre de bacterias, incluso la alimentación estéril; un laboratorio con protocolo de manejo de muestras de pacientes en protocolos de tratamiento específico y el manejo de muestras especiales (preparación de concentrados de leucocitos en la citometría hemática buffy coat y lograr una lectura óptima); área de preparación de medicamentos de alta especialización, y por otro lado, lo más importante es el personal de enfermería, auxiliar y administrativo, que con todo el grupo se esperan los resultados que tienen en otros países.
6. Trasplante de médula ósea (MO). Es un tipo de tratamiento complejo y de alto costo, que requiere de un donador de MO compatible y de un padecimiento inactivo con alta probabilidad de recaída temprana o tardía o, con factores de mal pronóstico. Es un procedimiento con tendencias más curativas ya que utiliza megadosis de quimioterapia para erradicar a las células leucémicas, pero en el intento, también erradica a las precursoras normales y se hace necesaria la reposición de una nueva médula normal compatible. El tipo alogénico (hermano compatible idéntico) es la mejor selección ya que el receptor lo acepta tan solo por identidad parcial (los únicos de aceptación total son los gemelos idénticos) y por tanto produce un rechazo del injerto contra el huésped, el que a su vez produce injerto contra leucemia y se incrementa la erradicación de la clona leucémica, no obstante, en casos graves del síndrome (grados 3-4) la morbimortalidad se incrementa a pesar del manejo específico.
LEUCEMIAS CRÓNICAS
1. Leucemia linfocítica crónica. Ocurre con más frecuencia en las personas de mayor edad y el criterio es la persistencia de de linfocitosis de más de 10 x 109/l, y la MO con infiltración de más del 50% de linfocitos con fenotipo CD5+. El criterio de administrar tratamiento es la duplicación de la cuenta de linfocitos en un año o la progresión de adenomegalia o esplenomegalia, aunque algunos casos escapan de este criterio por la presencia de anemia hemolítica o trombocitopenia autoinmune y entonces se indica el tratamiento a base de la combinación de fludarabina, ciclofosfamida y prednisona, en los estadios I y II sólo somenter a observación y esperar evolución sin tratamiento.
2. Leucemia mieloide crónica (LMC). En esta enfermedad existe un gran avance en el conocimiento de la presencia del cromosoma Filadelfia, que fue descrito en 1950 en esa ciudad de la Unión Americana y que a sus inicios significó el primer marcador cromosómico en asociación a malignidad, sin embargo al paso de los años de investigación, se logró conocer la t(9;22) con la expresión funcional del cromosoma con la producción de una oncoproteína con gran actividad de tirocinocinasa, que incrementa la proliferación celular y que a su vez explica la gran leucocitosis y trombocitosis con que se presentan estos pacientes, así como la gran esplenomegalia (figura 5).
Por estas fechas se cumplen de 10 a 12 años del descubrimiento de una pequeña molécula dirigida específicamente contra ese sustrato molecular donador de fosfatos para la regulación interna de la célula leucémica y sus sustratos. El inicialmente llamado STI (signal transduction inhibitor) produjo inhibición competitiva de la fosforilación, llevó a la célula a la apoptosis y los pacientes lograron resultados clínicos nunca antes vistos con remisiones moleculares de hasta el 80-90% a 10 años, lo que cambió dramáticamente la historia natural de la enfermedad, que con el tratamiento previo no era mayor a 3 años. Esta información sí hace un cambio en la historia natural y el pronóstico de la enfermedad, antes mortal a corto plazo.

Estos ejemplos son una base del avance tan intenso que se produjo en los últimos años y que es deseable que permita provocar el interés de los estudiantes de medicina, de sus profesores, pero sobre todo de las autoridades educativas de que la leucemia en general ocupa los primeros 5 lugares de frecuencia de las enfermedades malignas del adulto y de los primeros lugares en los niños, por lo que la hematología se deberá incluir dentro de las materias básicas en el curriculum de la carrera de Medicina y aún como parte de los cursos de especialización de posgrado.
Aunque es útil por su simpleza, la clasificación franco-americano británica (FAB) puede llevar a errores diagnósticos —y por lo tanto terapéuticos— hasta en el 20% de los casos. Por este motivo, la clasificación por métodos de inmunohistoquímica y biología molecular se ha convertido en un requisito sine qua non para la correcta clasificación y posterior manejo de los pacientes (figura 6).

A diferencia de la clasificación FAB, la de la OMS (tabla 2) refleja un cambio en el paradigma a través del cual entendemos a las enfermedades hemáticas, pues por primera vez se conjuntaron la información genética, las características morfológicas, citoquímicas e inmunofenotípicas con los hallazgos clínicos dentro de los algoritmos diagnósticos de las neoplasias del tejido hematopoyético; la importancia relativa de cada criterio difiere entre neoplasias y no existe un "estándar de oro" para la clasificación de todas las enfermedades hematológicas malignas. El objetivo fue definir entidades que pudieran ser reconocidas por los patólogos y que tuvieran relevancia clínica. Desde su aparición en 2001 se han realizado diferentes revisiones para actualizar su contenido en relación a los descubrimientos más actuales. La última revisión es del 2008 y clasifica a las neoplasias malignas hematológicas de la siguiente manera:
1. Neoplasias mieloides.
2. Neoplasias linfoides.
3. Enfermedades de los mastocitos o células cebadas.
4. Enfermedades histiocíticas y de células dendríticas.

NEOPLASIS MIELOIDES
Las neoplasias mieloides se derivan de progenitores en la médula ósea, que se diferencian en eritrocitos, granulocitos (neutrófilos, basófilos y eosinófilos), monocitos y megacariocitos. En la clasificación FAB se reconocen 3 categorías principales:
1. Leucemia mieloide aguda.
2. Síndromes mielodisplásicos.
3. Neoplasias mieloproliferativas.
Los determinantes más importantes de las categorías reconocidas utilizan sus características morfológicas, histoquímicas e inmunofenotípicas y son el porcentaje de blastos, el linaje celular y el grado de diferenciación de las células neoplásicas (figura 7). En años recientes, las características genéticas (citogenéticas y moleculares), así como el tratamiento previo y la evolución de la mielodisplasia, mostraron un impacto significativo en el comportamiento clínico de estos padecimientos que no siempre se correlacionan adecuadamente con las categorías de la FAB, por lo que el debate central para su reclasificación fue el discriminar entre las entidades patológicas y los factores pronósticos, para lograr una clasificación con relevancia clínica y significancia para el patólogo. Algunas anormalidades genéticas parecen definir a diferentes enfermedades, mientras que otras representan factores pronósticos de una enfermedad específica.

Actualmente, la clasificación de la OMS reúne a los padecimientos mieloides en 4 grupos principales:
1. Enfermedades mieloproliferativas
2. Síndromes mielodisplásicos
3. Enfermedades mielodisplásicas/mieloproliferativas
4. Leucemias agudas mieloides

Las enfermedades mieloproliferativas, son un grupo de trastornos clonales asociados con la proliferación de una o más líneas mieloides. Cada vez resulta más claro que estas enfermedades, se asocian a menudo con mutaciones que ocasionan incrementos anormales en la actividad de tirosino-cinasas y en la proliferación de células progenitoras de la médula ósea, independientes de factores de crecimiento. El porcentaje de blastos en médula ósea es normal o ligeramente elevado, pero siempre es menor al 20%. La hematopoyesis generalmente es efectiva, lo cual resulta en un incremento en las cuentas de una o más células maduras en sangre periférica. El prototipo de las neoplasias mieloproliferativas es la leucemia mieloide crónica cromosoma Filadelfia positivo (Ph1) (BCR/ABL). Las otras entidades que se incluyen son:
1. Policitemia vera.
2. Mielofibrosis idiopática.
3. Trombocitemia esencial primaria.
4. Leucemia eosinofílica crónica.
5. Leucemia neutrofílica crónica.
6. Mastocitosis.
7. Neoplasias mieloproliferativas no clasificables.

Los síndromes mielodisplásicos se refieren a trastornos que se caracterizan por una producción celular ineficaz y displasia, con un riesgo variable de transformación en leucemia aguda. La celularidad en la médula a menudo está incrementada pero es muy variable. Existe maduración pero también displasia de una o más líneas mieloides. La hematopoyesis no es efectiva y por lo tanto existen citopenias. En este rubro se incluyen:
1. Citopenia refractaria con displasia* de una línea.
• Anemia refractaria.
• Neutropenia refractaria.
• Trombocitopenia refractaria.
2. Anemia refractaria con sideroblastos anulares.
3. Citopenia refractaria con displasia de múltiples linajes.
4. Anemia refractaria con exceso de blastos.
5. Síndrome mielodisplásico con d(5q).
6. Síndrome mielodispásico inclasificable.
7. Síndrome mielodisplásico juvenil, incluye una entidad provisional conocida como citopenia refractaria juvenil.
Los síndromes mielodisplásicos/mieloproliferativos incluyen trastornos en los cuales coexisten características displásicas y proliferativas. En este grupo se icluye a la leucemia juvenil mielomonocítica, la cual es representativa de ambos síndromes (mielodisplásico y mieloproliferativo). Casi la mitad de los pacientes se presenta con cuentas de neutrófilos normales o bajas y displasia de múltiples líneas celulares sin organomegalia y médula ósea con morfología que semeja a la anemia refractaria con exceso de blastos, pero con monocitosis. Otros pacientes presentan neutrofilia intensa, monocitosis y esplenomegalia. Aún no se sabe si son 2 enfermedades diferentes, una mielodisplásica y una mieloproliferativa; sin embargo hasta el momento no existen diferencias en las anormalidades citogenéticas ni en los patrones de crecimiento de las colonias in vitro o en su evolución clínica, por lo que existe controversia entre clínicos y patólogos de acuerdo a su lugar dentro de la clasificación. De acuerdo a la última revisión, en este rubro se ubican:
1. Leucemia mielomonocítica crónica.
2. Leucemia mieloide crónica atípica (negativa para BCR/ABL).
3. Leucemia mielomonocítica juvenil.
4. Síndrome mielodisplásico/mieloproliferativo no clasificable.
En la categoría de las leucemias agudas mieloblásticas (LAM) (la cual se define por un porcentaje mayor al 20% de mieloblastos en la médula ósea o en sagre periférica, o la presencia de una anormalidad citogenética en particular a pesar de la cuenta de blastos) se reconoce a los siguientes grupos:
1. LAM con traslocaciones citogenéticas recurrentes.
2. LAM con características mielodisplásicas.
3. LAM y SMD relacionados con tratamientos antineoplásicos.
4. LAM no clasificable.
5. Sarcoma mieloide.
6. Proliferaciones mieloides relacionadas al síndrome de Down.
7. Neoplasia blástica plasmocitoide de células dendríticas.
NEOPLASIAS LINFOIDES
Son aquellas que se originan de las células que normalmente se desarrollan en linfocitos T (LT citotóxicos, colaboradores o reguladores) o linfocitos B (linfocitos o células plasmáticas). En general, las neoplasias linfoides se dividen en aquellas que derivan de precursores linfoides y aquellas provenientes de linfocitos maduros y células plasmáticas y posteriormente se agrupan de acuerdo a su estirpe (B o T).
Históricamente, las neoplasias linfoides que se presentan en la médula ósea y que involucran a la médula ósea se han separado de aquellas que se presentan como un tumor (linfoma). Sin embargo, ahora se sabe que cualquier linfoma se puede presentar con características clínicas de leucemia y que cualquier leucemia puede presentarse ocasionalmente como un tumor (sarcoma granulocítico). En la clasificación de la OMS, el diagnóstico de varias neoplasias linfoides depende no sólo de la localización anatómica de las células tumorales, sino del origen de la célula tumoral definido morfológicamente. Dichas consideraciones anularon la relevancia de los términos L1 y L2 de la clasificación FAB, pues no correlacionan con su inmunofenotipo, anormalidades genéticas o con su curso clínico (figura 8). La L3 es equivalente al linfoma de Burkitt en fase leucémica y se debe diagnosticar como tal.

1. Neoplasias de precursores. Existe el consenso de que las neoplasias de precursores que se presentan como tumores sólidos y aquellos que involucran a la médula ósea y la sangre son biológicamente la misma enfermedad con diferentes presentaciones clínicas. La mayoría de las neoplasias de precursores linfoides se presentan como leucemias, por lo que se acordó que la clasificación debería mantener el término LAL para la fase leucémica de las neoplasias precursoras de tipos B y T. existen 2 categorías principales:
• Leucemias/linfomas precursoras B.
• Leucemias/linfomas precursoras T.
2. Neoplasias de células B maduras. La clasificación propuesta considera a los linfomas y leucemias del mismo tipo celular como una misma enfermedad con diferentes presentaciones clínicas o estadios. Las enfermedades específicas derivadas de las células B maduras son las siguientes:
1. Leucemia linfocítica crónica/linfoma de linfocitos pequeños.
2. Linfoma linfoplasmacítico.
3. Linfoma de células del manto.
4. Leucemia prolinfocítica de células B.
5. Linfoma folicular.
6. Linfoma difuso de células grandes B.
• Linfoma intravascular de células grandes B.
• Linfoma mediastinal primario de células grandes B.
• Linfoma de células grandes B (relacionado con el virus de Epstein Barr-VEB).
• Linfoma de células grandes B rico en histiocitos y células T.
• Linfoma difuso de células grandes B del sistema nervioso central.
• Linfoma difuso de células grandes B cutáneo primario.
• Linfoma difuso de células grandes B del anciano positivo para VEB.
• Linfoma pasmablástico.
• Linfoma pleural primario.
• Linfoma de células grandes B positivo para alkoma (ALK).
• Linfoma de Burkitt.
7. Linfoma de células B de la zona marginal.
8. Linfoma de células B de la zona marginal extranodal.
9. Linfoma de células B de la zona marginal esplénico
10. Leucemia de células peludas.
11. Plasmocitoma/mieloma de células plasmáticas.

NEOPLASIAS DE LÍNEAS MIELOIDES Y LINFOIDES
Algunas neoplasias expresan marcadores tanto de líneas mielodes como linfoides, éste grupo representa a las leucemias de linaje ambiguo, que son aquellas que o no presentan características de línea linfoide ni mieloide (leucemia aguda indiferenciada) o presentan características de ambas líneas (leucemia aguda de fenotipo mixto o de líneas mixtas).
Tabla 3.Clasificación de la OMS de las neoplasias mieloides y leucemias agudas
BIBLIOGRAFÍA
Bassan R, Hoelzer D. Modern Therapy of Acute Lymphoblastic Leukemia. J Clin Oncol. 2011;29:523-43. [ Links ]
Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in Acute Leukemia. J Clin Oncol. 2011;29:487-94. [ Links ]
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011 May 12;117(19):5019-32. Epub 2011 Feb 7. Review. [ Links ]
Cortes J, Hochhaus A, Hugues T, Kantajian H. Front Line and Salvage Therapies with Tyrosine Kinase Inhibitors and other Treatments in Chronic Myeloid Leukemia. J Clin Oncol. 2011;29:524-31. [ Links ]
Chin-Hon Pui, Carroll WL, et al. Biology, Risk Stratification, and Therapy of Pediatric Acute Leukemia: An Update. J Clin Oncol. 2011;29:551-65. [ Links ]
Gambacorti PC, Antolin L, Hurtado MR. Multicenter independent assessment of ourcomes in chronic myeloid leukemia patients treated with Imatinib. Journal National Cancer Institute. 2011;103:1-9. [ Links ]
Grever M, Lozanki G. Modern Strategies for Hairy Cell Leukemia. J Clin Oncol. 2011:29;583-90. [ Links ]
Gribben JG, O'Brien S. Update on Therapy of Chronic Lymphocytic Leukemia. J Clin Oncol. 2011;29:544-50. [ Links ]
Hurtado MR, Vargas VP, Cortes FJ. Chronic Myeloid Leukemia. Current Concepts in Physiopathology and Treatment. Cancerología. 2007:2:137-47. [ Links ]
Hurtado MR, Vargas VP, et al. Imatinib compared with Imatinib/Cytarabine for the first-line treatment of early Philadelphia chromosome positive Chronic Myeloid leukemia. Results of a Randomized clinical trial of the Mexican Collaborative Leukemia Group. Clinical Leukemia. 2008: 2(2);1128-32. [ Links ]
Lichtman MA. Classification and clinical manifestations of the clonal myeloid disorders. En: Williams. Hematology. Mc Graw-Hill; 2010. [ Links ]
Marcucci G, Haferlach T, Dohner H. Molecular Genetics of adult acute myeloid Leukemia. Prognostic and Therapeutic Implications. J Clin Oncol. 2011;29:475-86. [ Links ]
Rafael Hurtado M Mellado Y, Floresw RG, Pablo Vargas. Semiología de la Citometría Hemática. Rev Fac Med UNAM. 2010; 53:36-43. [ Links ]
Sanz M, Lo-Coco F. Modern Approaches to treating Acute Promyelocytic Leukemia. J Clin Oncol. 2011;29:495-503. [ Links ]
* Displasia. Se refiere a la alteración citomorfológica que incluye disociación de la maduración núcleo-citoplasma (recuerde que la maduración de la cromatina depende del DNA y el citoplasma del RNA, por lo tanto el núcleo se detiene en la maduración mientras que el citoplasma sigue su proceso normal) lo cual produce células no viables y hay apoptosis intramedular.

