











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El proyecto del genoma humano derivó en una enorme cantidad de información genética, debido fundamentalmente al desarrollo tecnológico de la secuenciación de los ácidos nucleicos.1 Lo anterior permitió la implementación de la medicina de precisión, con el desarrollo de diferentes métodos moleculares diagnósticos y terapias blanco, dirigidas a mutaciones o variantes patogénicas.1,2
A la fecha son varios los ejemplos exitosos: las mutaciones se usan como marcadores moleculares, así como para el desarrollo tecnológico de novedosas drogas específicas o nuevas taxonomías oncogenómicas. Otros ejemplos son el estudio de la leucemia mieloide crónica relacionada con el cromosoma Filadelfia3 y del cáncer mamario con los genes supresores BRCA1/2.4 Una estrategia mucho más compleja que las anteriores es la prueba TMB (tumor mutational burden), con la que se buscan variantes patogénicas en más de 400 genes, para individualizar el manejo terapéutico enfocado a genes master o drivers.5,6 De igual forma ocurre con el cáncer medular de tiroides (CMT), ya que gracias a su asociación con variantes patogénicas en el oncogén RET, este es aceptado como otra prueba molecular en cáncer.7
No cabe duda de que la aplicación de técnicas ómicas-moleculares son estrategias fundamentales para identificar blancos terapéuticos que se adapten exclusivamente al individuo, por lo que las posibilidades son casi inimaginables.6
El gen RET y su proteína
El protooncogen RET contiene 21 exones y codifica para una proteína transmembranal de 1114 aminoácidos; en su región extracelular presenta cuatro dominios tipo cadherina y uno rico en cisteínas, lo que facilita la dimerización del receptor para su activación; en la región transmembranal, yuxtamembranal e intracelular posee dos dominios de actividad de tirosina cinasa (Figura 1).7,8
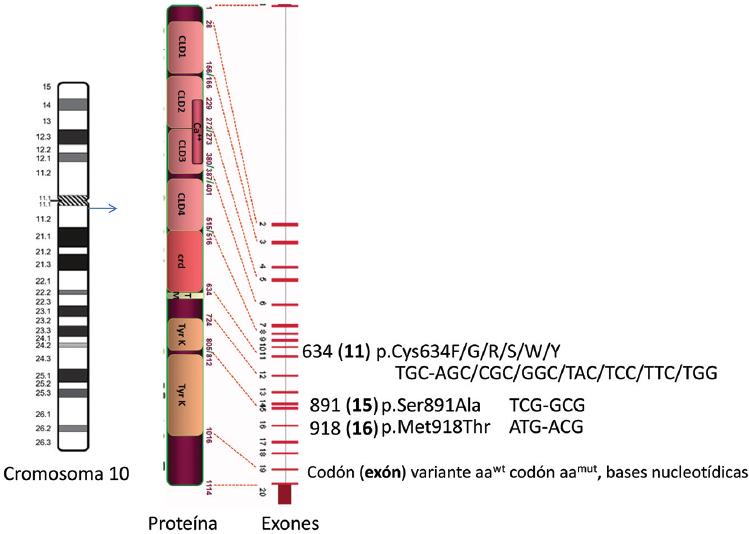
Figura 1 Esquema representativo del gen RET y su proteína. Ideograma del cromosoma 10 con el patrón de bandas G, la flecha indica la posición citogenética aproximada de RET. A la derecha, esquema representativo de la proteína RET que muestra las distintas regiones proteicas y los sitios de mutación correspondientes en los exones (líneas rojas pequeñas). En seguida se observan tres ejemplos de la nomenclatura para las variantes patogénicas de NEM2 y NEM3. Figura tomada y modificada de http://atlasgeneticsoncology.org/index.html.
En las etapas tempranas del desarrollo, RET induce el crecimiento y morfogénesis del sistema genitourinario, se expresa en la cresta neural y es esencial para el desarrollo del sistema nervioso entérico.9,10 En los adultos actúa como proteína de supervivencia de neuronas centrales y periféricas y se expresa en células C tiroideas.
Por su importante papel en la morfogénesis y maduración, no es sorprendente el papel de RET en diversas patologías:9 RET está mutado en el cáncer papilar del tiroides9,11 y sobreexpresado en enfermedades hematológicas9 y en carcinoma de páncreas, pulmón y mama.12-14
Del gen RET al CMT y neoplasias endocrinas múltiples (NEM)
La figura patognomónica del CMT son las células C parafoliculares alteradas.15 En términos genéticos, el CMT se presenta de manera autosómica dominante, por lo que 100 % de los sujetos con mutación en RET (heterocigotos) desarrollarán CMT en algún momento de su vida,16 el cual constituye una de las principales manifestaciones neoplásicas tempranas en la mayoría de los individuos NEM2 y 3.
Inicialmente, algunas familias NEM2 fueron clasificadas incorrectamente con CMT familiar (CMTF), aspecto de interés clínico ya que es indispensable descartar oportunamente alteraciones en las glándulas paratiroides, feocromocitomas, etcétera, y definir que los CMTF son pacientes NEM2 verdaderos.15,17,18 En este contexto y acorde a la Asociación Americana de Tiroides, la agresividad clínica del CMT depende de la variante patogénica en RET, conocimiento que ha permitido formar grupos de riesgo (Tabla 1).16 Independientemente del tipo de variantes patogénicas presentes, estas incrementan la vida media de la proteína.
Tabla 1 Recomendaciones para testeo molecular de variantes patogénicas de RET y tiroidectomía profiláctica
| Categoría de riesgo ATA para el CMT | Codón en el gen RET (exón) | Edad indicada para el testeo molecular* | Edad indicada para tiroidectomía profiláctica** |
|---|---|---|---|
| Muy alto | 918 (16) | — | Uno a seis meses |
| Alto | 634, 883 (11, 15) | A los tres años | Antes de los 5 años |
| Moderado | 533, 609, 611, 618, 620, 630, 631, 666, 768, 790, 804, 891, 912 (10, 11, 13-16) | A los cinco años | Infancia o adolescencia con base en la revisión de los niveles de calcitonina en suero |
Las palabras resaltadas con negritas indican el exón donde se localizan los codones de sitios de mutación.
*Exploración física anual, ecografía de cuello y medición de calcitonina sérica.
**Los pacientes deben excluirse antes de la tiroidectomía si ya presentan feocromocitoma. Estas recomendaciones son acordes con las de la Asociación Americana de Tiroides (ATA, American Thyroid Association).16
Otro dato clínico de importancia es que en las familias con CMT se presenta el fenómeno de anticipación: al heredar la mutación, el fenotipo se presentará aproximadamente 10 años más temprano que en el padre o madre afectado, es decir, en cada generación el individuo afectado es más joven.17
El reto es encontrar la variante patogénica en RET. Ya que estas mutaciones se localizan en sitios calientes, su búsqueda y detección se facilita tecnológicamente en los familiares (Figura 1). Estos sitios se localizan dentro de los exones 5, 8, 10, 11, 13-16, virtualmente en 100 % de los casos, y son característicos para el diagnóstico de CMTF, CMT esporádico o NEM2 y NEM3.11,15,16
Correlación del genotipo-fenotipo de RET
Los estudios genéticos sobre las mutaciones en RET indican una fuerte correlación genotipo-fenotipo, por lo que esta relación es un arma poderosa y su valor radica en la predicción del inicio de la enfermedad y el pronóstico; actualmente se utiliza como una guía para la intervención terapéutica y el manejo de los pacientes.
La complejidad de RET se aprecia en la diversidad de sus variantes; bases de datos como ARUP o COSMIC reportan 199, 44.8 % patogénicas, 51.4 % de significado incierto y el resto de índole benigna.19-21
Más de 95 % de los pacientes con NEM2 presenta una de las variantes patogénicas en los codones 609, 611, 618, 620, 630 o 634,9,22 mientras NEM3 virtualmente se asocia al codón 918. Este síndrome se caracteriza por neuromas mucosos, habitus marfanoide, CMT, feocromocitoma, así como ganglioneuromatosis gastrointestinal, sin hiperparatiroidismo. Las primeras manifestaciones se observan a edad más temprana que en los pacientes con NEM2, frecuentemente durante la lactancia, y suelen acompañarse de estreñimiento crónico, distensión abdominal, diarrea o megacolon al nacimiento.22 Las proteínas originadas por esas variantes se asocian a un fenotipo severo y de peor pronóstico cuya expectativa de vida no excede de los 20 años.9
Diagnóstico clínico-molecular de CMT/NEM
El CMT se presenta frecuentemente con la detección fortuita (incidental) o por palpación de un nódulo tiroideo aislado no doloroso o por el crecimiento de un ganglio cervical. El ultrasonido y la citología por aspiración con aguja fina confirman el diagnóstico y se procede a realizar la tiroidectomía total con disección selectiva de ambos cuellos por la agresividad biológica de esta neoplasia.6,16,22,23
La metodología de apoyo diagnóstica utilizada en México para los CMT/NEM se establece con la detección de los niveles séricos de calcitonina ≥ 10 pg/mL y del antígeno carcinoembrionario.16
En un esfuerzo para brindar una atención médica integral, durante las últimas dos décadas nuestro equipo de investigación está incluyendo la detección de mutaciones de RET. Para ofrecer un abordaje integral y decidir la conducta a seguir, la prueba molecular se realiza en células sanguíneas y tumorales, para discriminar la mutación en línea germinal o somática.15,17
El cribado bioquímico en los familiares de un paciente CMT/NEM2-NEM3 se realiza con el método estándar de determinación de calcitonina, frecuentemente tras la estimulación con pentagastrina; por el contrario, el cribado molecular se realiza una sola vez en la vida.
La detección temprana permite reducir la morbimortalidad, puesto que una pronta tiroidectomía es curativa en determinados estadios.24,25 De tal forma, resulta claro que la determinación bioquímica combinada con el testeo genético-molecular aumenta la sensibilidad y agudeza del proceso de tamizaje. En nuestra experiencia, el testeo molecular solo resulta de enorme utilidad, mientras que una vez detectada la mutación, el paciente es sometido a la determinación de calcitonina con notable costo-beneficio.
Asesoramiento y medicina de precisión
Una vez que los pacientes tienen el diagnóstico clínico, altos niveles séricos de calcitonina y el gen mutado, deben recibir asesoramiento genético debido a la predisposición de su descendencia a desarrollar la enfermedad. En tal caso, todos los familiares de primer grado del probando deben someterse al diagnóstico molecular de RET. En este contexto, dependiendo del grado de riesgo por la mutación, está indicada la tiroidectomía profiláctica o la disección de los ganglios linfáticos centrales.26
No obstante, antes de diseñar planes de tratamiento quirúrgico deben sopesarse los beneficios oncológicos que se esperan de la operación contra los riesgos. En el beneficio neto de la cirugía profiláctica debe considerarse el riesgo continuo de morbilidad oncológica si no se trata esta patología, la reducción del riesgo relativo del tratamiento (curación quirúrgica, probablemente alta) y el riesgo de daño del tratamiento (morbilidad posquirúrgica, probablemente baja en manos experimentadas).27 Acorde a lo anterior, hay dos escuelas para actuar éticamente en estos casos: realizar la cirugía hasta que el portador presente sintomatología o efectuar la cirugía profiláctica a la edad más temprana posible.22 La Asociación Americana de Tiroides (Tabla 1)16 recomienda que la tiroidectomía profiláctica se efectué durante el primer año e, incluso, durante los primeros meses de vida en niños que se encuentran en la categoría de muy alto riesgo (M918T). Los niños en la categoría de alto riesgo (C634F/G/R/S/W/Y, A883F) deben someterse a tiroidectomía profiláctica a la edad de cinco años, o antes en caso de que presenten niveles elevados de calcitonina basal o estimulada. La disección de los ganglios linfáticos centrales se puede aplicar a niños con calcitonina > 40 pg/mL o metástasis en los ganglios linfáticos detectada por métodos de imagen. En este caso, la experiencia quirúrgica del cirujano es determinante.26
En niños con riesgo de desarrollo de CMT, el diagnóstico genético precoz y la cirugía profiláctica temprana en manos experimentadas se asociaron a excelentes resultados entre siete y 16 años después del procedimiento. Cuando los niños eran mayores, 10 años en comparación con 6.1 años, en la tiroidectomía profiláctica solo 44/50 (88 %) se curaron bioquímicamente, en comparación con 114/115, 99.1 %. Estos datos apoyan que un adecuado asesoramiento y abordamiento diagnóstico de la enfermedad se relacionan con tasas de curación bioquímica de 100 %, ausencia de enfermedad estructural residual o recurrencia, y rara vez con alguna morbilidad operatoria permanente.27
El testeo molecular de RET en el Instituto Mexicano del Seguro Social
El equipo multidisciplinario de investigación genómica conformado por investigadores clínicos y biomédicos trabaja como Centro de Diagnóstico Mutacional de RET. Para su creación, fue necesaria la participación de líderes especialistas de otras entidades del sector salud, un esfuerzo sin precedentes. El Centro recopila la información para la generación de bases de datos y divulgación de la epidemiología de CMT-RET en la población mexicana.
Hasta la fecha, siete familias con CMT/NEM3 no relacionadas fueron positivas (ATG > ACG) a M918T somática y germinal; la mayoría de los sujetos fueron del sexo masculino (edad ≈ 15 años) con calcitonina > 400 pg/mL antes del procedimiento quirúrgico, con seguimiento por más de 100 meses. Una paciente femenina de 59 años con calcitonina sérica de 2400 pg/mL presentó la mutación somática; el seguimiento se prolongó por seis meses. Todos los pacientes fueron sujetos a procedimiento quirúrgico y presentaron niveles de calcitonina ≤ 10 pg/mL en controles posteriores. Histológicamente presentaron un patrón de CMT clásico y solo uno con patrón oncocítico.
Familias mexicanas con CMT/NEM2
Se han estudiado más de 20 familias CMT/NEM2 no relacionadas, la mayoría positivas para las variantes C634R, dos C634W y una C634T, todas con la histología de patrón clásico.
Especialmente de una familia con más de 20 años de seguimiento se obtuvo información clínica valiosa; se trató de 50 miembros estudiados que correspondieron a cinco generaciones. Inicialmente fue diagnosticada como CMTF, con cinco miembros C634R (somática y germinal) positivos. Por presentar la mutación germinal, todos los integrantes con vínculos sanguíneos (primos, hermanos, tíos, sobrinos, hijos) fueron sujetos al testeo molecular. De esta forma, se identificaron 15 portadores de la mutación, cinco menores de tres años quienes fueron sujetos a tiroidectomía profiláctica. Las piezas quirúrgicas mostraron hiperplasia de paratiroides. En el seguimiento aproximado de 16 años, todos reportan curación bioquímica, ausencia de enfermedad residual y ninguna evidencia de recurrencia (datos en proceso para publicación), resultados que concuerdan con los de una revisión europea. A los de ocho años del diagnóstico inicial, el probando debutó con feocromocitoma, por lo cual esta familia se clasificó como NEM2. Con el análisis genético se logró identificar que la variante C634R se presenta con alta penetrancia, 79 % a la edad de 30 años.17 Con estos resultados sugerimos que los pacientes positivos para dicha variante deberían ser reclasificados como NEM2, tal como sucede con la variante C618S.28 Esto proporciona evidencia de que las variantes patogénicas 618, 634 o 918 están asociadas a NEM2 o NEM3, respectivamente.
Los datos anteriores harán posible el desarrollo de técnicas diagnósticas de nueva generación y adecuaciones para sistemas simples, precisos, específicos, flexibles y económicos, con el fin de generar programas y algoritmos accesibles a todos los sectores de la población (Figura 2).15,29-31
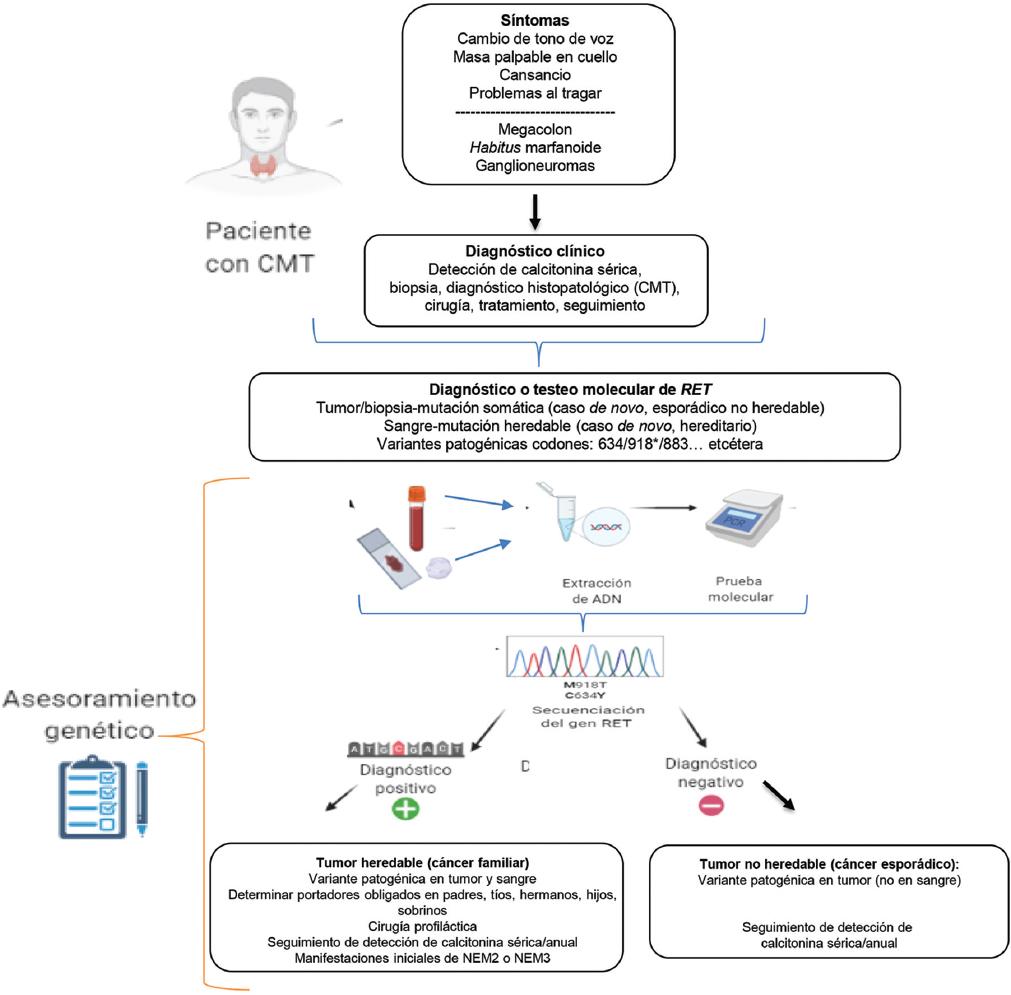
Figura 2 Algoritmo sugerido en el diagnóstico molecular del gen RET. Se muestra la estrategia para el testeo molecular en la detección de variantes patogénicas del gen RET. En el recuadro se observan los principales síntomas sugerentes de CMT, NEM2 y NEM3. Estos síndromes se descartan con el diagnóstico clínico. En caso positivo se procede al testeo molecular. La detección de la mutación se debe realizar a partir de una muestra de sangre (germinal) y de tejido (o incluido en parafina, somático). De las muestras se extrae el ADN, el cual se somete a amplificación por reacción en cadena de la polimerasa (PCR). Posteriormente se realiza la secuenciación del ADN amplificado, con lo que se genera el electroferograma, como se muestra en la figura. La prueba positiva en sangre es indicativa de mutación germinal o del tipo heredable, por lo que se debe efectuar un tamizaje con esa misma mutación en cada uno de los familiares del probando; en los sujetos positivos se sugiere asesoramiento genético y quirúrgico. El resultado negativo para mutación en sangre y tejido es indicativo de cáncer no asociado a RET y de cualquier otro tipo de mutación en otros genes.
Afortunadamente, los síndromes CMT/NEM2-NEM3 son patologías poco frecuentes32,33 que se relacionan etnográficamente y cuyas mutaciones se siguen transmitiendo en la actualidad.34 Lo anterior se demuestra por los escasos datos publicados, que representan un lapsus por la rareza de la frecuencia de estos síndromes, o bien, por las tendencias de la ciencia,35-37 de ahí que se exhorte a incrementar los estudios sobre estas patologías.
Conclusiones
La medicina de precisión en México es una realidad. Como resultado de la revolución molecular y genómica, la detección oportuna de las enfermedades genera la posibilidad de realizar intervenciones para prevenir la aparición de síntomas o minimizar su gravedad.
El conocimiento sobre los cambios genéticos de cada persona tiene como objetivo ayudar a decidir cuál tratamiento será el adecuado o puede tener mayor probabilidad de funcionar en un paciente específico.38,39

