











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
En 1905, Langley introdujo la idea de receptor o sustancia receptiva al observar la competencia entre diversos venenos. Esta idea permanece como concepto hasta avanzado el siglo XX, cuando se define su estructura proteica y se identifican los genes que los codifican. Es importante destacar que existen varias grandes familias de receptores:
– Receptores intranucleares.
– Receptores canal.
– Receptores con actividad enzimática o que se asocian a enzimas.
– Receptores acoplados a proteínas G, motivo de esta revisión y a los que se indicará como GPCR (G protein-coupled receptors).
Estas familias de receptores no están relacionadas filogenéticamente.
Receptores acoplados a proteínas G y su acción
Los receptores acoplados a proteínas G se denominan así ya que ejercen muchas de sus acciones al interaccionar con GTPasas heterotriméricas (Figura 1), que a su vez modulan la actividad de enzimas (Figura 1) y también de algunos canales iónicos. Robert J. Lefkowitz, médico especializado en cardiología, ha sido pionero y pieza fundamental en el estudio de estos receptores, por lo que recibió el Premio Nobel de Química en 2012.1
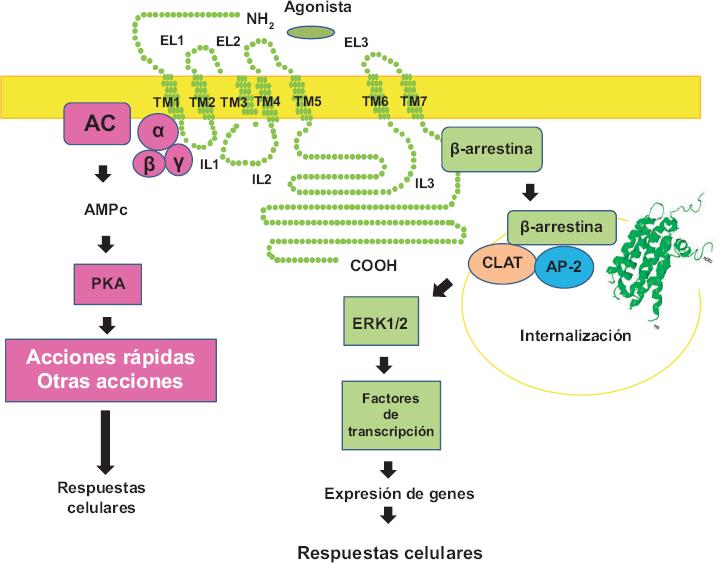
Figura 1 GPCR extendido en la membrana plasmática con sus siete dominios transmembranales (TM 1-7), sus asas extracelulares (EL 1-3) e intracelulares (IL 1-3), así como sus extremos amino (NH2) y carboxilo (COOH) terminales. También la imagen cristalográfica de un GPCR (endocitado). En la porción izquierda se muestra la señalización a través de la proteína G (α, β y γ) y el sistema de la adenil ciclasa (AC) que genera AMP cíclico (AMPc) y activa a la proteína cinasa A (PKA). En la parte izquierda se indica la señalización endosomal a través de la β-arrestina hacia las MAP cinasas (ERK 1/2). AP-2: proteína adaptadora, CLAT: clatrina.
Es importante señalar que existen varias familias de proteínas G:
– Gs, asociadas a la activación de la adenil ciclasa.
– Gi, asociadas a la inhibición de la adenil ciclasa y a la modulación de diversos canales iónicos.
– Gq/11, asociadas a la generación de IP3 y diacilglicerol, con la movilización de calcio intracelular y la activación de la proteína cinasa C.
– G12/13, asociadas a la GTPasa rho y a la dinámica del citoesqueleto, fundamentales para determinar la forma celular y la migración.2
Si bien algunos GPCR tienen una fuerte preferencia para interaccionar con un solo tipo de proteína G, otros muchos pueden acoplarse a varias.
En el humano se conocen aproximadamente 800 genes que codifican para GPCR, muchos de ellos con ligandos naturales conocidos y otros en los que aún no se conoce el ligando endógeno (receptores huérfanos). Dada su enorme variedad e importancia, no es sorprendente que se haya estimado que entre 30 y 40 % de los fármacos de uso común en la práctica médica tienen su blanco en los GPCR,3 al igual que numerosas drogas ilegales.
En la Figura 1 se ilustra esquemáticamente un GPCR en su forma extendida a lo largo de la membrana y la estructura real cristalográfica en el receptor internalizado. Como puede apreciarse en la forma extendida, estos receptores constan de siete zonas que atraviesan la membrana plasmática, por lo que reciben también el nombre de receptores de siete dominios transmembranales. Los segmentos transmembranales se unen entre sí mediante tres asas intracelulares y tres asas extracelulares; el extremo aminoterminal se localiza en el exterior de la célula, mientras que el extremo carboxilo es intracelular. También se muestra que el receptor ejerce su acción primariamente desde la membrana plasmática a través de activar a las proteínas G. Además, se indica que puede existir otra señalización a través de la proteína β-arrestina (principalmente detectada en los receptores internalizados, es decir, que han migrado, por tráfico vesicular, a los endosomas).4,5 Esta proteína tiene la capacidad de unirse a una gran cantidad de elementos de señalización.6 En la misma Figura 1 se ilustra el proceso más estudiado, la activación de las MAP (mitogen activated protein), cinasas que conducen a la expresión genética.
Durante la desensibilización, los receptores se fosforilan, lo que favorece su unión a las β-arrestinas que bloquea la señalización a través de las proteínas G. Posteriormente se descubrió que lo que ocurre es un cambio de un tipo de señal (mediado por las proteínas G) a otro (mediado por las β-arrestinas). La fosforilación del receptor puede ser por la presencia del agonista de este (frecuentemente catalizada por una familia de cinasas de GPCR, denominadas GRK) o a través de la activación de otros receptores por la activación de diversas proteínas cinasas en sus cascadas de señalización (Figura 2).

Figura 2 Los GPCR pueden ser fosforilados (P), en respuesta a su activación, por cinasas de GPCR (GRK) u otras proteínas cinasas (PK) activadas por las cascadas de acción de diferentes receptores (se muestran un GPCR y un receptor con actividad de tirosina cinasa). En la parte inferior se muestra la consecuencia funcional más frecuente (desensibilización).
Hasta hace relativamente pocos años, se consideraba que los receptores permanecían inactivos hasta que se producía un estímulo. Los agentes que activan al receptor se denominaban agonistas y los que bloqueaban la acción, antagonistas. Este concepto se ha modificado los últimos años. En primer lugar, se ha observado que numerosos receptores tienen cierta actividad en ausencia de ligando, denominada actividad constitutiva, y que hay agentes que la disminuye, ahora llamados agonistas inversos (Figura 3, panel A). Hay agentes que incrementan la actividad hasta el máximo, denominados agonistas totales, en tanto otros no llegan al máximo efecto aun cuando aumente su concentración, los cuales reciben el nombre de agonistas parciales (Figura 2, panel A). Esto se debe a que pueden existir varias conformaciones activas del receptor; algunos agentes inducen una configuración óptima (agonistas totales), mientras que otros logran conformaciones subóptimas, pero suficientes para inducir cierto efecto (agonistas parciales). Nótese que hablamos de eficacia; la potencia es otro concepto y se refiere a la concentración del agente capaz de inducir la mitad de su efecto máximo.

Figura 3 Diferentes tipos farmacodinámicos de acción. A: efecto de diferentes tipos de agentes sobre la actividad de los GPCR. B: diferentes agonistas pueden manifestar “sesgo” hacia acciones mediadas por las proteínas G o hacia acciones mediadas por la β-arrestina (C).
En la Figura 3 (panel A), el agonista parcial es menos eficaz, pero más potente que el agonista total.7 Los antagonistas puros o “agonistas neutros” son agentes que interaccionan con el receptor, pero no modifican su actividad. Los agonistas inversos reducen la actividad basal del receptor y la respuesta biológica. Esto es más fácil de observar en sistemas modelo en los que se sobreexpresa al receptor de interés, que en tejidos u órganos, en los cuales la densidad de receptores es habitualmente baja y frecuentemente se expresan diversos tipos de receptores para un agente dado, lo que complica el estudio. Una gran cantidad de los agentes que denominamos antagonistas son en realidad agonistas parciales de baja actividad intrínseca o agonistas inversos. Es importante recordar que estos son términos operativos y que un solo agente puede ser agonista total para una acción, parcial para otra e incluso agonista inverso para una tercera (ver más adelante).
Estos compuestos tienen su acción sobre el “sitio ortostérico” del receptor, es decir, en algunos de los residuos con los que los agonistas naturales se unen. Otros agentes actúan en sitios diferentes, incluso distantes, llamados “alostéricos” y que pueden tener acción positiva o negativa sobre la acción de los agonistas. Si bien esto es novedoso para los GPCR, no lo es para otros receptores como los receptores canal. Un ejemplo clásico son las benzodiacepinas, moduladores alostéricos de los receptores canal de los receptores para el GABA tipo A.8
Otro aspecto de gran importancia ilustrado en la Figura 3 (paneles B y C) es el de los agonistas sesgados. Se denominan así dado que presentan un sesgo o preferencia en su acción, es decir, son eficaces o potentes para desencadenar una acción más que otra. En el caso ilustrado, se indica que un agente prefiere las acciones inducidas a través de las proteínas G, mientras que el otro es mejor para las mediadas a través de las β-arrestinas. Sin embargo, esta no es la única posibilidad para mostrar sesgos. Por ejemplo, un agonista puede favorecer una configuración en el receptor que le permita tener una muy buena acción sobre una proteína G y no sobre otra. Es igualmente posible que un agente favorezca la internalización y degradación de un tipo de receptor, lo que lo convertiría en un agonista de corto plazo, pero comportarse como un “antagonista funcional” (al depletar a la célula de receptores) a mediano y largo plazo. Las posibilidades que se abren son muchas y hay evidencia experimental para algunas de ellas. Más adelante se dan algunos ejemplos de interés médico. Es evidente que la clasificación farmacodinámica aún está en evolución.
Estructura de los GPCR. Activación y unión con la proteína G
En el año 2000, por cristalografía de rayos X se analizó el primer GPCR cristalizado, la rodopsina, el receptor de la luz.9 A la fecha, diversos GPCR se han analizado por esta técnica y por criomicroscopia electrónica: los receptores β2-adrenérgicos,10,11 de cannabinoides CB112 y de endotelina B1,13 así como el LPA1 del ácido lisofosfatídico14 y el receptor μ de los opioides,15 entre otros.
Brian Kobilka, también médico cardiólogo, recibió el Premio Nobel de Química 2012, junto con su asesor de posdoctorado, Robert Lefkowitz. Su trabajo fue pionero en la cristalización de los GPCR y el análisis de los cambios estructurales que presentan en presencia de agonistas totales (receptor activo) y agonistas inversos (receptor inactivo); además, logró la cristalización del receptor acoplado a la proteína Gs10,11,16 (véase conferencia del Premio Nobel en https://www.nobelprize.org/prizes/chemistry/2012/kobilka/lecture/), lo que ha permitido acercarse a los eventos moleculares que ocurren durante la activación del complejo ligando-GPCR-proteína G. El análisis cristalográfico, en conjunto con el modelado molecular in silico, se han convertido en herramientas importantes para definir los mecanismos de acción de diferentes agentes y el diseño molecular de fármacos.
Los GPCR como blancos terapéuticos
Los opiáceos, particularmente la morfina, la heroína y el fentanilo, son compuestos que se caracterizan por su efecto analgésico y causan en los pacientes una sensación de bienestar general. Desafortunadamente, son fuertemente adictivos y en forma rápida causan tolerancia y dependencia. El uso hedonístico ilegal se ha vuelto común, lo que ha generado una crisis en muchos países. Los efectos indeseables más observados por la estimulación prolongada de los receptores m de los opiáceos incluyen náuseas, vómito, constipación gastrointestinal y grave depresión respiratoria que puede llevar al paciente a la muerte.
Existe cierta evidencia que sugiere que la mayoría de los efectos benéficos de los opiáceos se pudieran deber a la activación de las proteínas G, mientras que los indeseables estarían asociados con la activación de las β-arrestinas. Por ello, se han diseñado diversos fármacos con efectos sesgados con la intención de evitar los efectos indeseables.17 Entre ellos se encuentra el oliceridine, un potente y eficaz activador de las proteínas G, pero un débil agente para inducir la fosforilación del receptor, su internalización y la activación de la β-arrestina; en roedores ha demostrado ser un buen analgésico que induce poca depresión respiratoria.18 Sin embargo, los estudios clínicos, aún iniciales, indican que es capaz de inducir depresión respiratoria y otros efectos indeseables. Lo mismo se ha observado con otros agentes desarrollados hasta el momento, lo que indica que si bien estos agonistas sesgados son promisorios, aún distan de ser una realidad clínica.17,19
Los receptores β1 y β2-adrenérgicos son de los más estudiados por su notable efecto sobre el funcionamiento cardiovascular. Los receptores β1-adrenérgicos son muy abundantes en el corazón, pero es claro que en el corazón humano existen también receptores β2-adrenérgicos y que estos tienen un papel importante en el funcionamiento normal del corazón y en diversas cardiopatías. El receptor β2-adrenérgico es el mejor estudiado. Además, los agonistas y antagonistas β-adrenérgicos son empleados en la clínica en forma cotidiana, tanto en los servicios de urgencias y terapia intensiva, como en el paciente ambulatorio que padece enfermedades crónicas como hipertensión arterial, insuficiencia cardiaca congestiva crónica y asma, entre otras.
Se conoce que bajo la acción de diferentes agentes, los receptores β2-adrenérgicos adquieren diferentes conformaciones y que ello parece estar relacionado con su acción y su utilidad clínica.20 En la Figura 4 se presenta un análisis de modelado de la interacción del receptor β2-adrenérgico con diversos ligandos. Como puede apreciarse, la noradrenalina, el agonista natural, hace contacto con residuos del receptor que pertenecen a las zonas transmembranales 3, 5 y 7, mientras que el alprenolol, el carvedilol y el bucindolol tienen menos contacto con la zona transmembranal 5, pero mayor con residuos de la zona transmembranal 6; interacciones diferentes se ven con los otros agentes, lo que sugiere que el receptor adquiere conformaciones distintas. Está reportado que el carvedilol, un “antagonista” no selectivo para los receptores β1 y β2-adrenérgicos, presenta una acción como “agonista sesgado” (nótense las dificultades para la clasificación farmacodinámica) sobre el receptor β2-adrenérgico hacia la vía de la β-arrestina y está indicado para el tratamiento de la insuficiencia cardiaca congestiva crónica.21 Este compuesto forma enlaces con residuos de la tercera asa intracelular que son parte del “interruptor” de acoplamiento proteína G/β-arrestina. Además, datos muy recientes indican claramente que este compuesto tiene una interacción con la β-arrestina que involucra al receptor β2-adrenérgico y a la proteína Gi.22
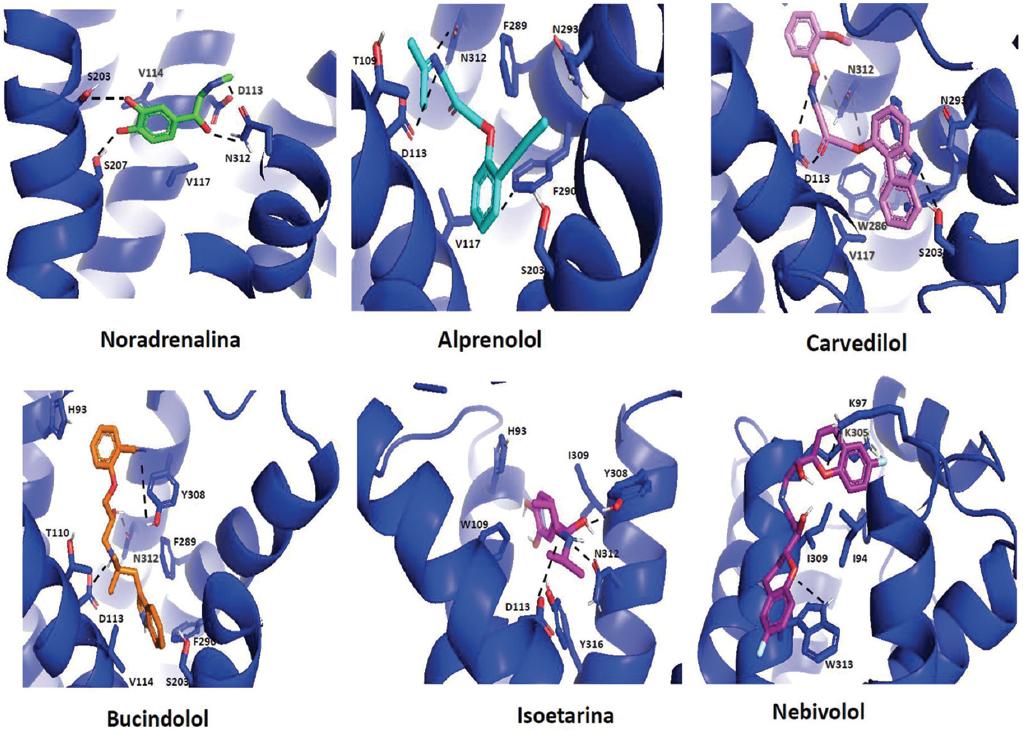
Figura 4 Análisis in silico de la unión de diferentes ligandos al receptor β2-adrenérgico. Los modelos se obtuvieron con el programa AutoDock Tools, disponible en internet (http://autodock.scripps.edu/resources/adt).
Conclusiones y discusión
El conocimiento de las vías de transducción de señales, la regulación de los GPCR por fosforilación y de la estructura de estos receptores ha sido de suma importancia en el avance del tratamiento de enfermedades en las que participan los GPCR. La obtención de agonistas sesgados que modulan y estabilizan la conformación del receptor, ya sea para activar a la proteína G o reclutar a la β-arrestina, parece ser clave para el tratamiento de múltiples enfermedades. El conocimiento del detalle estructural de los distintos estados de un GPCR unido a su agonista es esencial para entender el mecanismo de activación del receptor y para el diseño y optimización de los ligandos.

