











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
La hemofilia es un trastorno hemorrágico hereditario provocado por la deficiencia cuantitativa del factor VIII (FVIII) de la coagulación, denominada hemofilia A (HA), la cual representa el 80% de los casos; o del factor IX (FIX), conocida como hemofilia B (HB), que corresponde al 20% restante. La deficiencia de estos factores ocasiona una incapacidad en la generación de trombina y amplificación de la fase fluida de la coagulación, con la subsecuente diátesis hemorrágica en las personas con hemofilia (PcH). Las manifestaciones clínicas de la HA y la HB son iguales y dependen de la cantidad de factor deficiente circulante, con el principal sitio de sangrado a nivel articular (hemartrosis) en los casos severos, el cual sin una terapia adecuada e integral puede desarrollar una artropatía hemofílica crónica, lo que representa la principal causa de morbilidad en esta población. El patrón de herencia es recesivo ligado al sexo (cromosoma X), por lo que los varones manifiestan la enfermedad y las mujeres son portadoras asintomáticas o con mínimos síntomas hemorrágicos. La prevalencia y alteraciones genéticas de la hemofilia son semejantes mundialmente, sin influir la ascendencia u origen étnico.
El pilar del tratamiento de la PcH es la infusión intravenosa (IV) del factor deficiente, ya sea a demanda (durante episodios hemorrágicos) o de manera profiláctica (administración del factor de forma regular) con el objetivo principal de evitar las hemartrosis espontáneas, sin embargo, con un riesgo considerable del desarrollo de inhibidores en hemofilia severa.
El objetivo del presente documento es realizar una revisión y consenso por parte de los médicos tratantes de hemofilia en una de Las principales instituciones de salud en México, describiendo desde los aspectos básicos de genética, fisiopatología y diagnóstico de la hemofilia; los esquemas de tratamiento, las principales complicaciones y las situaciones especiales asociadas con el incremento de la supervivencia de la PcH. Por último, se analiza la hemofilia adquirida, donde la fisiopatología, características clínicas y manejo difieren significativamente de la hemofilia congénita.
Antecedentes históricos y epidemiología
La descripción de un problema hemorrágico ligado al sexo es reconocido desde el siglo II a. C. plasmado en el Talmud1, acerca de los cuidados en los varones recién nacidos al ser circuncidados si existía el antecedente de hemorragia en hermanos varones. En el siglo xvii se describe como «la enfermedad de la realeza», al ser transmitida por la reina Victoria a los descendientes de los tronos de Gran Bretaña, Alemania y España. El caso más reconocido afectó a Alexei, hijo de Alexandra (nieta de la reina Victoria) y el último zar de Rusia, Nicolás II2. En 1803 John Conrad Otto, médico de Filadelfia, publicó el primer artículo reconociendo un padecimiento hemorrágico familiar que afectaba principalmente a hombres. El término de hemofilia, del griego hemo «sangre» y filia «amor», se publicó en 1824 por Hopff. En 1947, el Dr. Alfredo Pavlosky, de Buenos Aires (Argentina), distinguió los dos tipos de hemofilia hereditaria.3
La incidencia mundial de HA es de 1/5,000 varones nacidos y de 1 en 30,000 para HB. Actualmente se calcula que existen 400,000 PcH en el mundo. El primer reporte mundial con información demográfica, de tratamiento y complicaciones de las PcH fue publicado en 1999 por la Federación Mundial de Hemofilia (en delante WFH, por sus siglas en inglés: World Federation Hemophilia) bajo la denominación de Annual Global Survey, que en la última publicación con datos del 2018, informó de un total de 210,454 PcH, con una distribución por severidad de acuerdo al ingreso económico, que se describe en la tabla 1. En México se reportaron un total de 5,814 casos: 4,761 con hemofilia A y 724 con hemofilia B, con un predominio de población mayor de 19 años (Tablas 2 y 3) y sin definición del tipo de hemofilia en 329 casos.4
Tabla 1 Distribución por severidad de hemofilia en países con ingreso económico medio-alto
| Severidad | ||||
|---|---|---|---|---|
| Leve | Moderada | Severa | Desconocida | |
| Hemofilia A | 19.49% | 23.26% | 41.82% | 15.44% |
| Hemofilia B | 20.04% | 28.67% | 36.42% | 14.86% |
Fuente: Annual Global Survey, 2018.4
Tabla 2 Distribución por edad de pacientes con hemofilia A
| Hemofília A | 0-4 | .5-13 | 14-18 | 19-44 | 45+ | Edad desconocida | |
|---|---|---|---|---|---|---|---|
| Francia | 6446 | 7% | 15% | 10% | 40% | 28% | 0% |
| Georgia | 270 | 3% | 21% | 6% | 49% | 21% | 0% |
| Ghana | 278 | 10% | 45% | 21% | 14% | 0% | 10% |
| Grecia | 815 | 4% | 8% | 7% | 46% | 34% | 0% |
| Guatemala | 250 | 4% | 17% | 16% | 44% | 9% | 10% |
| Guyana | 13 | 0% | 15% | 8% | 62% | 15% | 0% |
| Honduras | 299 | 5% | 27% | 14% | 43% | 3% | 7% |
| Hong Kong | 119 | 3% | 24% | 8% | 55% | 10% | 5% |
| Hungría | 914 | 4% | 3% | 5% | 35% | 42% | 11% |
| India | 17,606 | 2% | 14% | 10% | 38% | 8% | 28% |
| Indonesia | 2035 | 11% | 32% | 16% | 36% | 3% | 1% |
| Irán | 5208 | 4% | 13% | 8% | 56% | 19% | 0% |
| Irak | 1867 | 23% | 40% | 20% | 15% | 3% | 0% |
| Irlanda | 715 | 14% | 16% | 8% | 33% | 29% | 0% |
| Israel | 608 | 10% | 17% | 10% | 39% | 24% | 0% |
| Italia | 4135 | 3% | 9% | 6% | 40% | 42% | 0% |
| Jamaica | 43 | 5% | 12% | 21% | 33% | 26% | 5% |
| Kenia | 504 | 12% | 29% | 27% | 14% | 13% | 4% |
| Corea | 1721 | 5% | 11% | 8% | 54% | 22% | 0% |
| Kirguistán | 343 | 7% | 38% | 14% | 32% | 8% | 0% |
| Líbano | 169 | 10% | 14 | 14% | 45% | 16% | 1% |
| Lesoto | 24 | 4% | 8 | 63% | 25% | 0% | 0% |
| Lituania | 152 | 0% | 0 | 0% | 0% | 0% | 100% |
| Madagascar | 70 | 11% | 36 | 11% | 39% | 3% | 0% |
| Malawi | 35 | 6% | 31 | 6% | 20% | 0% | 37% |
| Malasia | 900 | 10% | 19 | 11% | 41% | 10% | 10% |
| Maldivas | 14 | 14% | 29 | 7% | 29% | 21% | 0% |
| Mali | 128 | 20% | 48 | 16% | 14% | 0% | 2% |
| Mauritania | 61 | 7% | 48 | 21% | 20% | 5% | 0% |
| Mauricio | 74 | 0% | 9 | 12% | 39% | 28% | 11% |
| México | 4761 | 3% | 20 | 11% | 42% | 10% | 14% |
| Mongolia | 81 | 17% | 40 | 9% | 30% | 5% | 0% |
| Montenegro | 41 | 2% | 15 | 12% | 32% | 39% | 0% |
| Marruecos | 183 | 11% | 36 | 8% | 41% | 4% | 0% |
| Birmania | 464 | 22% | 36 | 13% | 20% | 3% | 7% |
| Nepal | 562 | 13% | 19 | 18% | 36% | 7% | 6% |
| Países Bajos | 797 | 6% | 10 | 9% | 32% | 43% | 0% |
| Nueva Zelanda | 450 | 4% | 11 | 13% | 36% | 35% | 0% |
Fuente: Annual Global Survey, 2018.4
Tabla 3 Distribución por edad de pacientes con hemofilia B
| Hemofília B | 0-4 | .5-13 | 14-18 | 19-44 | 45+ | Edad desconocida | |
|---|---|---|---|---|---|---|---|
| Finlandia | 33 | 3% | 27% | 15% | 33% | 21% | 0% |
| Francia | 1498 | 7% | 17% | 11% | 37% | 28% | 0% |
| Georgia | 53 | 6% | 19% | 2% | 49% | 25% | 0% |
| Ghana | 16 | 13% | 25% | 25% | 0% | 0% | 38% |
| Grecia | 184 | 4% | 8% | 4% | 39% | 45% | 0% |
| Guatemala | 34 | 3% | 17% | 17% | 29% | 3% | 31% |
| Honduras | 34 | 12% | 24% | 18% | 38% | 3% | 6% |
| Hong Kong | 24 | 0% | 21% | 8% | 38% | 33% | 0% |
| Hungría | 230 | 2% | 1% | 4% | 38% | 43% | 12% |
| India | 2715 | 2% | 13% | 12% | 42% | 11% | 21% |
| Indonesia | 310 | 16% | 38% | 19% | 24% | 1% | 3% |
| Irán | 1109 | 3% | 12% | 8% | 59% | 18% | 0% |
| Irak | 502 | 23% | 40% | 20% | 13% | 5% | 0% |
| Irlanda | 243 | 4% | 14% | 11% | 41% | 30% | 0% |
| Israel | 104 | 14% | 15% | 18% | 37% | 15% | 0% |
| Italia | 886 | 3% | 10% | 9% | 42% | 37% | 0% |
| Jamaica | 4 | 0% | 0% | 25% | 50% | 25% | 0% |
| Kenia | 114 | 18% | 25% | 31% | 21% | 4% | 2% |
| Corea | 427 | 4% | 14% | 11% | 49% | 22% | 0% |
| Kirguistán | 40 | 8% | 15% | 15% | 58% | 5% | 0% |
| Líbano | 47 | 6% | 11% | 23% | 51% | 9% | 0% |
| Lituania | 24 | 0% | 0% | 0% | 0% | 0% | 100% |
| Madagascar | 57 | 16% | 46% | 16% | 23% | 0% | 0% |
| Malawi | 6 | 0% | 50% | 0% | 17% | 0% | 33% |
| Malasia | 172 | 8% | 17% | 15% | 41% | 13% | 6% |
| Maldivas | 4 | 50% | 25% | 25% | 0% | 0% | 0% |
| Mali | 13 | 62% | 15% | 8% | 15% | 0% | 0% |
| Mauritania | 15 | 7% | 47% | 20% | 20% | 7% | 0% |
| Mauricio | 13 | 8% | 8% | 23% | 46% | 8% | 8% |
| México | 724 | 5% | 19% | 11% | 44% | 10% | 12% |
| Mongolia | 28 | 11% | 39% | 18% | 25% | 7% | 0% |
| Montenegro | 4 | 0% | 0% | 25% | 50% | 25% | 0% |
| Marruecos | 34 | 10% | 29% | 23% | 39% | 0% | 0% |
| Birmania | 76 | 45% | 33% | 7% | 11% | 4% | 1% |
| Nepal | 93 | 8% | 30% | 18% | 27% | 11% | 6% |
| Países Bajos | 125 | 3% | 13% | 15% | 31% | 38% | 0% |
| Nueva Zelanda | 102 | 4% | 8% | 8% | 33% | 46% | 0% |
Fuente: Annual Global Survey, 2018.4
Fisiopatología
La hemofilia A y B son enfermedades hemorrágicas de carácter hereditario recesivo ligadas al sexo en el 70% de los casos (el resto de los casos son consecuencia de mutaciones espontáneas de novo), con afección prácticamente exclusiva del sexo masculino por su genotipo XY,1 debido a que los genes que codifican para los factores VIII y IX están localizados en el brazo largo del cromosoma cromosoma X que determina el sexo, en las posiciones Xq28 y Xq27, respectivamente.5 Se han descrito más de 4,000 variantes patogénicas en estos genes que causan una disminución cuantitativa-cualitativa en la expresión y actividad proteica de los factores de coagulación.6
Para fines didácticos los defectos genéticos del FVIII se pueden describir en tres grupos: 1) rearreglos genéticos, como la inversión del intrón 22, que se presenta en el 45% de los pacientes con hemofilia severa y es causada por recombinación homóloga entre la secuencia 9.5 kb y dos regiones homologas extragénicas, así como la inversión del intrón 1, que se presenta en el 1-2% de los casos severos; 2) inserciones o deleciones de secuencias genéticas, y 3) sustitución de una sola base de ADN que involucra las mutaciones de sentido falso (missense), sin sentido (nonsense) o cambios del marco de lectura (frameshift). En las figuras 1-3 se describen las alteraciones cromosómicas en HA dependiendo de su severidad, donde predomina la mutación puntual, seguida de las deleciones, duplicaciones e inserciones. En pacientes con HB las mutaciones puntuales se presentan en la mayoría de los casos, seguidas de las deleciones, inserciones y duplicaciones (Figs. 4-6).7
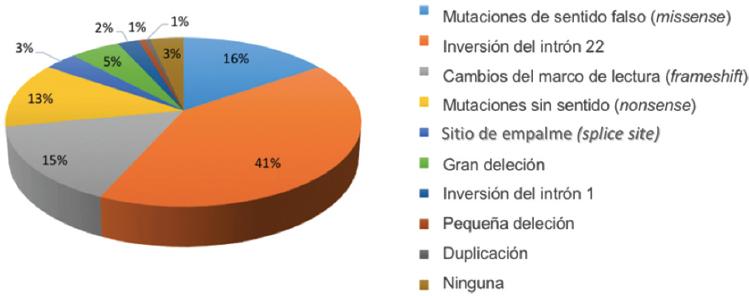
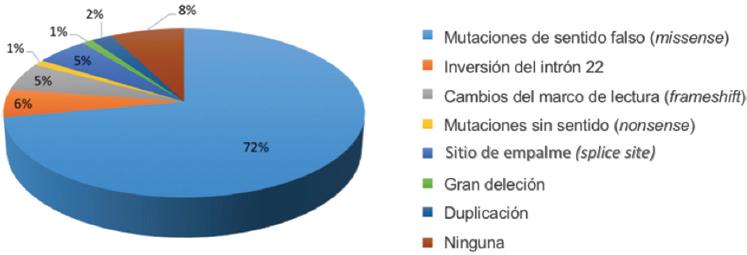


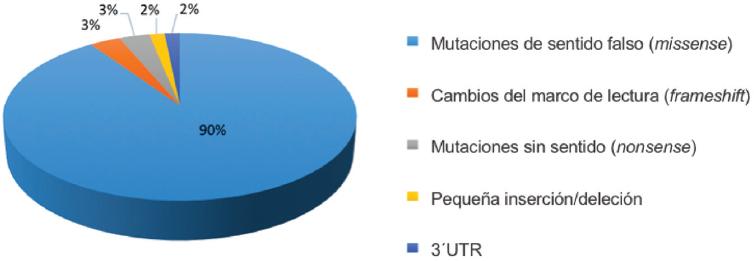
Figura 5 Mutaciones en hemofilia B moderada (adaptada de Miller, et al., 20117). 3´UTR: untranslated region (región no traducida de un gen).
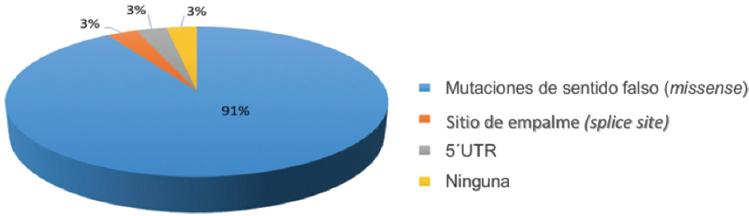
Figura 6 Mutaciones en hemofilia B leve (adaptada de Miller, et al., 20117). 5´UTR: untranslated region (región no traducida de un gen).
Patrón de herencia
En la figura 7 se señala el patrón de herencia clásico de hemofilia, donde se concluye que todas las hijas de un paciente hemofílico (XY) son portadoras obligadas al heredar el cromosoma X afectado, los descendientes varones serán sanos al recibir el cromosoma Y normal del padre (XY). Para el caso de mujeres portadoras (XX), en cada embarazo existe un riesgo del 25% de concebir un hijo varón afectado (hemofílico), un 25% de concebir una hija portadora y el 50% de probabilidad restante de tener un hijo o hija no afectados. En términos generales, el 50% de los descendientes varones y el 50% de las hijas de madres portadoras serán hemofílicos o portadoras, respectivamente.1
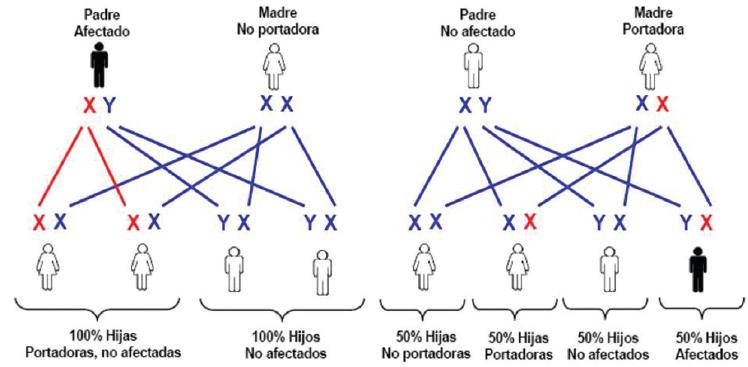
Figura 7 Patrón de herencia en hemofilia. Se señala en color rojo el cromosoma X afectado y en azul los cromosomas no afectados.
Desde el punto de vista genético, la mujer recibe doble información (paterna y materna) del cromosoma X. Durante el periodo embrionario se presenta una inactivación al azar de uno de los dos cromosomas X, fenómeno biológico conocido como lionización, de tal manera que las portadoras tendrán en promedio un 50% de células que expresan el alelo normal8 para la síntesis del factor implicado, cantidad suficiente para mantener el nivel del factor alrededor del 50% de actividad (rango: 22-116 UI/dl) y cursar asintomáticas;1 sin embargo, en ocasiones la lionización no es al azar y se produce una inactivación extrema del cromosoma X, lo que genera una doble población celular en donde la mayoría de las células somáticas tienen inactivado el cromosoma X con el gen normal, ocasionando una menor expresión del factor de coagulación y generando el concepto de «portadoras con niveles bajos», con manifestaciones hemorrágicas semejantes a un hemofílico leve o moderado y/o menorragia. El mismo fenómeno de lionización del cromosoma X es el responsable de los niveles bajos de factor VIII o IX en mujeres con síndrome de Turner o mosaicismo del cromosoma X. Aunque la hemofilia es un padecimiento que se manifiesta en el varón hemicigótico y la mujer heterocigótica es portadora (ídem) y generalmente asintomática, en teoría una mujer puede ser hemofílica si el padre es hemofílico y la madre portadora.
Con los antecedentes genéticos mencionados es pertinente mencionar algunos aspectos relevantes de las portadoras de acuerdo con los antecedentes familiares. Se definen como portadoras obligadas a: a) las hijas de una PcH; b) la madre de un varón con hemofilia y con antecedentes familiares; c) la madre de un hemofílico y con un familiar portador del gen de hemofilia, y d) madres de dos o más varones con hemofilia. Las portadoras probables son: a) todas las hijas de una portadora; b) la madre de un hemofílico sin antecedente familiar de hemofilia, y c) hermanas, madres, abuelas maternas, tías, sobrinas y primas de portadoras obligadas.
Alteración de la hemostasia en hemofilia
La coagulopatía de la PcH es consecuencia de la incapacidad para magnificar, controlar y mantener la generación de trombina por la deficiencia del FVIII o el FIX. La generación de trombina se posiciona como un evento de alto valor biológico-fisiológico al ser parte esencial del complejo molecular responsable de la fase fluida de la hemostasia. Al presentarse una lesión tisular, el FIX activado (FIXa) se une al FVIII activado (FVIIIa) sobre una capa lipídica rica en factor tisular (FT) formando el complejo «diezasa intrínseco», el cual tiene la capacidad de generar el 90% de la trombina ante un daño tisular, con una eficiencia 106 veces mayor que los FVIII y FIX de manera aislada (Fig. 8). Este complejo supera en 50 veces al complejo «diezasa extrínseco» (con alto contenido de FVIIa) para la activación del FX en FXa, con la subsecuente activación del factor II (protrombina) en trombina, la cual convierte el fibrinógeno soluble (factor I) en fibrina (insoluble).1 Esta descripción sencilla de un segmento específico de la hemostasia explica cómo la ausencia de los factores VIII y IX se manifiesta clínicamente con eventos hemorrágicos clásicos de la PcH.

Diagnóstico y clasificación
Se debe sospechar de hemofilia en un varón que presenta un sangrado prolongado y excesivo, no relacionado con la magnitud del trauma y/o una hemorragia que se presenta horas después de la lesión o de tipo recidivante. En las pruebas de coagulación primarias, el número de plaquetas, el tiempo de protrombina (TP), el tiempo de trombina (TT) y el fibrinógeno serán normales, con un tiempo de tromboplastina parcial activado (TTPa) prolongado, lo cual se describe más delante. Las manifestaciones hemorrágicas de la PcH A o B son indistinguibles desde el punto de vista clínico, por lo que es necesario identificar el factor deficiente para la reposición específica de este. El diagnóstico definitivo se basa en la cuantificación de los factores de coagulación. La Organización Mundial de la Salud definió que una unidad internacional (UI) es la actividad del factor presente en 1 ml de plasma y dependiendo de la nomenclatura de cada lugar se puede expresar de manera equivalente como: 1 UI/dl, 0.01 UI/ml o 1%. La severidad de la hemofilia se clasifica de acuerdo con la actividad del nivel plasmático circulante sin tratamiento de FVIII o FIX en severa, moderada o leve (Tabla 4).9
Tabla 4 Clasificación de hemofilia y correlación de manifestaciones hemorrágicas
| Severidad | Nivel de factor de coagulación | Episodios de hemorragia |
|---|---|---|
| Severa | <1 UI/dl (< 0.01 UI ml) o < 1% | Sangrados espontáneos en articulaciones o músculos |
| Moderada | 1-5 UI/dl (0.01-0.05 UI ml) o 1-5% | Sangrado espontáneo ocasional; sangrados prolongados con trauma menor o cirugía |
| Leve | 5-40 UI/dl (0.05-0.40 UI ml) o 5-40% | Sangrado severo con trauma o cirugía mayor. El sangrado espontáneo es raro |
Adaptada de Blanchette, et al., 2015.9
Estudios de laboratorio
Los estudios de hemostasia tienen un papel fundamental en el diagnóstico y seguimiento de la PcH. El aseguramiento de calidad de estas pruebas incluye el control de calidad interno y externo, así como el control de los factores que pueden influir en las diferentes etapas del procesamiento de las pruebas, como la fase preanalítica, donde ocurren más del 70% de los errores de laboratorio (requisición de exámenes realizada por el médico, registro correcto del estudio solicitado, preparación, recolección y toma de muestra). Lo anterior tiene relevancia al considerar que las pruebas de coagulación son excepcionalmente susceptibles a los cambios de temperatura, en particular por la termolabilidad del FVIII.
A continuación se especifican los aspectos relevantes en el procesamiento de estudios de laboratorio de la PcH y una breve descripción de los hallazgos en los estudios de tamizaje, confirmatorios y para detección de inhibidores en hemofilia.10-12
ASPECTOS GENERALES
- Venopunción: asegurar una muestra atraumática con uso mínimo del torniquete (aplicar justo antes de la extracción), utilizando agujas de calibre 19 a 21 G (calibre 23 G en pacientes pediátricos).
- Tubo de colección con anticoagulante citrato de sodio al 3.2%: debe llenarse con al menos el 90% de lo indicado (proporción 9:1 entre la muestra obtenida y el anticoagulante).
- Mezclar completamente la muestra con el anticoagulante invirtiendo la punta del tubo de 4 a 6 veces de manera gentil y asegurar que no se formen coágulos.
- Transporte de muestras: a temperatura ambiente y centrifugación en la primera hora de obtención, en caso de traslado a un laboratorio de preferencia congelar el plasma inmediatamente a 20 °C o menos y transportarse con hielo seco.
- Ayuno: no es obligatorio, aunque un exceso de lípidos puede afectar a los analizadores analíticos.
ESTUDIOS DE TAMIZAJE ANTE SOSPECHA DE HEMOFILIA
- Biometría hemática: dentro de los parámetros de referencia si no existe otra alteración justificable.
- TP normal y TTPa prolongado.
- Corrección con plasma: en hemofilia congénita el TTPa se corregirá mezclando el plasma del paciente en una relación 1:1 con plasma normal. Si la mezcla no corrige el TTPa prolongado puede indicar la presencia de un inhibidor o presencia de anticoagulante en el plasma.
ESTUDIOS CONFIRMATORIOS DE DOSIFICACIÓN DE FACTORES VIII Y IX
La determinación del FVIII se puede realizar por ensayo cromogénico o coagulométrico. La dosificación del FIX se determina mediante ensayo coagulométrico de una etapa. Se recomienda realizar una dosificación integral de todos los factores que pueden prolongar el TTPa (VIII, IX, XI y XII) durante la evaluación inicial. Cuando existe el antecedente familiar de hemofilia se puede determinar la actividad del FVIII o IX en sangre del cordón umbilical de los recién nacidos varones.
Recomendación: en pacientes sin antecedentes hereditarios de hemofilia, con cuadro clínico hemorrágico y TTPa prolongado, realizar correcciones con plasma y confirmar la actividad del factor de coagulación deficiente mediante ensayo cromogénico o coagulométrico para FVIII y coagulométrico para FIX. En los casos con antecedente familiar de hemofilia realizar la búsqueda intencionada con determinación del factor de coagulación específico en sangre de cordón umbilical o sangre periférica del recién nacido.
DETECCIÓN DE ANTICUERPOS (INHIBIDORES) ANTI-FACTOR VIII Y ANTI-FACTOR IX
Los anticuerpos anti-FVIII o anti-FIX son aloanticuerpos de tipo inmunoglobulina (Ig) G con actividad neutralizante (inhibidor) o no neutralizante de la actividad del factor de coagulación y representan una complicación grave de la terapia sustitutiva con concentrado de factor de coagulación (CFC), por lo que son más frecuentes en PcH severa. Se deben sospechar en pacientes con respuesta clínica inadecuada a la administración del factor deficiente, particularmente si había respondido con anterioridad y/o un cambio en el fenotipo hemorrágico.12
La confirmación de inhibidor y la cuantificación del título se realiza mediante el ensayo Bethesda o su modificación Bethesda-Nijmegen, este último con mayor sensibilidad y especificidad, el cual consiste en mezclar volúmenes iguales del plasma problema con plasma normal, incubación a 37 °C por 2 horas y medir la actividad residual del factor en la mezcla, utilizando como control un plasma libre de factor VIII o IX. Por definición, una unidad Bethesda (UB) es el título de inhibidor que neutraliza el 50% de actividad del factor en un mililitro de plasma.
Si después de la incubación el factor residual equivale al 100% del nivel en la muestra control, entonces el nivel de inhibidor es cero. Si el FVIII residual equivale al 50 o al 25% del control, el nivel de inhibidor es de 1 o 2 UB, respectivamente (Fig. 9). En caso de resultado menor al 25%, el plasma del paciente se somete a diversos grados de dilución hasta que el resultado pueda leerse en la gráfica y el resultado se multiplica por el grado de dilución para expresarlo en UB. Por ejemplo, si una mezcla de plasma se diluye 1:5 antes de incubación y el factor residual es 50%, o una unidad, 1 x 5 = 5 UB.13,14
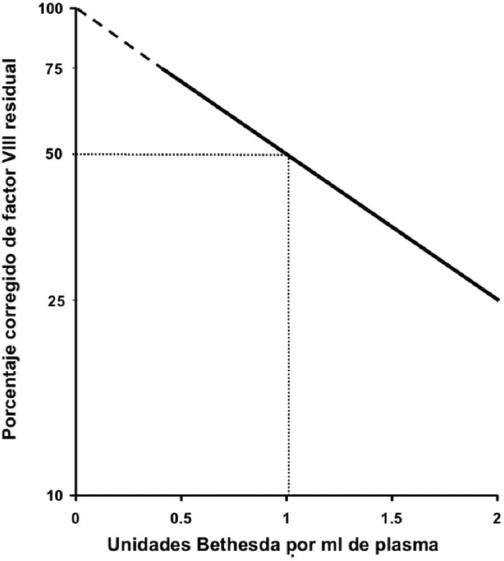
Figura 9 Correlación entre factor VIII residual y unidades Bethesda (tomada de Diagnóstico y tratamiento de inhibidores de los factores VIII y IX, 200413).
Recomendación: realizar la búsqueda de inhibidores mediante la técnica de Bethesda-Nijmegen en los pacientes sin exposición previa al factor de coagulación (PUP, por sus siglas en inglés: previously untreated patients) a los 20, 50, 100 y 150 días de exposición (DE; se define como DE a la infusión de FVIII o IX administrada en 24 h).
En pacientes previamente tratados con factores de coagulación (en delante PTP, por sus siglas en inglés: previously treated patients) realizar la búsqueda de inhibidor en caso de cambios en el fenotipo hemorrágico y previo a cirugías programadas.
Diagnóstico diferencial
Una vez documentada la disminución de FVIII las posibles causas son: HA congénita, Hemofilia A adquirida (HAA) o enfermedad de von Willebrand (EvW) tipo 2N, por lo que se debe realizar un diagnóstico diferencial entre estas patologías, considerando las características clínicas del patrón hemorrágico, edad de presentación, antecedentes hereditarios, resultados de estudios básicos de coagulación (Tabla 5) y panel de EvW (por lo menos antígeno y multímeros de von Willebrand, y cofactor de ristocetina).
Tabla 5 Diagnóstico diferencial de deficiencia de FVIII por estudios básicos de hemostasia
| Posible diagnóstico | TP | TTPa | TS | Plaquetas |
|---|---|---|---|---|
| Hemofilia A o B | Normal | Prolongado | Normal | Normal |
| EvW | Normal | Normal o prolongado | Normal o prolongado | Normal o disminuido |
| Hemofilia adquirida | Normal | Prolongado | Normal | Normal |
TP: tiempo de protrombina; TTPa: tiempo de tromboplastina parcial activado; TS: tiempo de sangrado; EvW: enfermedad de von Willebrand. Adaptada de Srivastava, 202012.
Diagnóstico genético
La información genética de la PcH representa una herramienta útil para predecir el riesgo del desarrollo de inhibidores y facilita la asesoría prenatal en portadoras.5 Para las PcH A los primeros estudios de tamizaje genético son la búsqueda de inversión del intrón 22 y el intrón 1; en caso de no detectarse alguna de estas estas alteraciones se realiza la secuenciación completa del gen del FVIII. Para la HB se realiza la secuenciación de los ocho exones del gen del FIX para la detección de mutaciones o deleciones.10
El estudio genético de portadoras puede ser complejo, en estos casos la secuenciación de los genes del FVIII y IX es el estudio de elección que permite detectar el rearreglo genético con una confiabilidad del 99%.15 Alrededor del 80% de las madres de un caso esporádico pueden ser portadoras de una mutación y en el 20% restante de los casos no se detecta alguna mutación y puede ser secundario a mosaicismo.
El diagnóstico prenatal es parte integral de los cuidados a las portadoras de hemofilia y es de relevancia para la culminación del embarazo. Los estudios incluyen técnicas no invasivas para el producto, como son la determinación del sexo del producto mediante el análisis del ADN fetal en sangre materna (factible en el primer trimestre del embarazo) o por ultrasonido desde las 15 semanas de edad gestacional (SEG), los cuales no son concluyentes, o bien, mediante métodos invasivos como el muestreo de las vellosidades coriónicas entre las 10-12 SEG o la amniocentesis, la cual se puede realizar entre las semanas 15 a 18 de embarazo. Si el cariotipo es 46 XY se pueden realizar los estudios genéticos del DNA extraido. La cordocentesis (muestra del cordón umbilical percutánea), que se emplea entre las 18-21 SEG es útil para determinación de FVIII y/o IX.16
Recomendación: aquellos centros que dispongan de los recursos diagnósticos deben de realizar el perfil genético de la PcH, iniciando con la búsqueda de la inversión 1 y la 22 para el caso de HA, en caso negativo realizar la secuenciación completa del gen del FVIII. Para HB realizar la secuenciación del gen del FIX, así como en las portadoras y/o el envío de muestras para protocolos de investigación.
Manifestaciones clínicas
El fenotipo clínico de la PcH es la tendencia a la hemorragia, lo cual se correlaciona con el nivel de actividad del factor de coagulación circulante (Tabla 4). En la PcH severa el 90% presenta un sangrado anormal en el primer año de vida. El principal sitio de sangrado ocurre en el sistema musculoesquelético, como se describe en la tabla 6. La hemartrosis es la principal manifestación hemorrágica en la PcH, se describe clínicamente como una sensación de «hormigueo» o «aura» en la articulación que precede a los signos clásicos de calor, edema, dolor y limitación del rango de movimiento. La presencia de tres o más sangrados espontáneos en una articulación en un periodo consecutivo de seis meses se define como «articulación blanco», la cual presenta cambios progresivos e irreversibles como deformación, limitación al movimiento y dolor crónico que pueden ameritar manejo quirúrgico e importante terapia de rehabilitación.
Tabla 6 Frecuencia aproximada de sangrados en diferentes sitios
| Sitio de sangrado | Frecuencia aproximada (%) |
|---|---|
| Hemartrosis | 70-80 |
| Más frecuente en articulaciones con movilidad: tobillos, rodillas y codos | |
| Menos frecuente en articulaciones multiaxiales: hombros, muñecas, caderas | |
| Múscular | 10-20 |
| Otros sangrados mayores | 5-10 |
| SNC | < 5 |
SNC: sistema nervioso central. Tomada de Srivastava, et al., 202012.
Toda hemorragia en la PcH es grave, sin embargo existen áreas anatómicas que ponen en riesgo la vida y ameritan de tratamiento inmediato (Tabla 7). La hemorragia intracraneal (HIC), ya sea espontánea o posterior a un traumatismo leve, es la principal causa de muerte, por lo que toda PcH y sospecha clínica de HIC debe tratarse con la aplicación inmediata de CFC antes de cualquier procedimiento diagnóstico. El sangrado en cuello o garganta y a nivel gastrointestinal representan también una urgencia en la PcH, por el riesgo de formación de un hematoma retrofaríngeo que pueda ocluir la vía aérea o de choque hipovolémico, respectivamente. En este punto es pertinente mencionar al hematoma del iliopsoas, caracterizado en sus formas graves por una hemorragia masiva retroperitoneal y manifestado con un dolor en ingle o fosa iliaca y una posición antálgica de flexión de cadera ipsilateral que en ocasiones es confundido con cuadros apendiculares, retrasando el manejo específico y favoreciendo las potenciales complicaciones.12
Tabla 7 Sitios de hemorragia en hemofilia
| Grave | Con peligro para la vida |
|---|---|
| Articular (hemartrosis) | Intracraneal |
| Múscular, especialmente compartimentos profundos (iliopsoas, pantorrilla y antebrazo) | Cuello/garganta |
| Mucosas (boca, encías, nariz y tracto genitourinario) | Gastrointestinal |
Tomada de Srivastava, et al., 202012.
Tratamiento
Manejo multidisciplinario
La atención adecuada de las diversas necesidades de la PcH y de su familia es mediante la intervención de un equipo multidisciplinario integrado por enfermería, psicología, nutrición, ortopedia, rehabilitación, estomatología, terapia ocupacional, trabajo social y genética, coordinados por el hematólogo y en apego a las guías nacionales de tratamiento. Todos los miembros del equipo deben tener experiencia y capacidad para tratar los trastornos de la coagulación y estar disponibles para atender a los pacientes en tiempo y forma. Deberá existir la infraestructura de un centro de tratamiento para hemofilia destinado a la atención de emergencias en todo momento, con acceso a estudios de laboratorio específicos (determinación de factores de coagulación e inhibidores), así como a los medicamentos y CFC necesarios.
El equipo multidisciplinario orientará a paciente y familiares sobre los síntomas precoces de una hemorragia con el objetivo de otorgar el tratamiento oportuno, y capacitará sobre la conservación, preparación y técnica de aplicación de los factores de coagulación, estableciendo un vínculo efectivo entre pacientes, familia y miembros del equipo de atención integral que promueva la adherencia al tratamiento, así como del cuidado de los accesos venosos en la PcH, ya que constituyen líneas de acceso vitales, mediante las siguientes recomendaciones:12,17
- Utilizar agujas tipo mariposa calibre 23 o 25 G.
- No realizar venodisección, a excepción de casos de emergencia.
- Después de una punción venosa, aplicar presión durante 3 a 5 minutos. Evitar en lo posible el uso de dispositivos de acceso venoso permanente, aunque puede ser necesario en casos específicos.
TRATAMIENTO FARMACOLÓGICO
El tratamiento farmacológico de primera línea en hemofilia es la aplicación del CFC deficiente, ya sea de tipo recombinante o derivado plasmático. Las opciones terapéuticas de aplicación son a demanda o de manera profiláctica, las cuales se exponen a continuación.
TRATAMIENTO A DEMANDA
Es la aplicación del CFC cuando existe evidencia clínica de una hemorragia aguda, calculando la dosis para incrementar la actividad del factor basada en la severidad de la hemorragia. El tratamiento a demanda ha demostrado disminuir la mortalidad y la progresión de la artropatía, pero no prevenirla.18,19 En hemorragias que ponen en peligro la vida, la dosis inicial del CFC debe proporcionarse de inmediato, aun antes de completar la evaluación diagnóstica inicial para obtener una actividad del 80 al 100%, mientras que en sangrados leves a moderados el objetivo es mantener una actividad de factor entre el 35 y el 50%. Las dosis de mantenimiento en HA generalmente se administran cada 12 horas, y cada 24 horas en la HB. Las dosis y duración del tratamiento con CFC dependerán del sitio, gravedad de la hemorragia y respuesta al tratamiento (Tabla 8).12,17
Tabla 8 Dosis inicial y de mantenimiento de CFC de acuerdo con el sitio de hemorragia
| Tipo de hemorragia | Actividad del factor % deseado HA | Dosis de factor VIII U/kg | Dosis de factor IX U/kg | Actividad del factor % deseado HB | Duración (días) |
|---|---|---|---|---|---|
| SNC/cabeza | |||||
| Inicial | 80-100 | 40-50 | 60-80 | 60-80 | 1 a 7 |
| Mantenimiento | 50 | 25 | 30 | 30 | 8 a 21 |
| Vía aérea (cuello) | |||||
| Inicial | 80-100 | 40-50 | 60-80 | 60-80 | 1 a 7 |
| Mantenimiento | 50 | 25 | 30 | 30 | 8 a 14 |
| Iliopsoas y muscular profunda con lesión NV | |||||
| Inicial | 80-100 | 40-50 | 60-80 | 60-80 | 1 a 2 |
| Mantenimiento | 30-60 | 15-30 | 30-50 | 30-50 | 3 a 5 Profilaxis secundaria durante fisioterapia |
| Cirugía mayor | |||||
| Preoperatorio | 80-100 | 40-50 | 60-80 | 60-80 | |
| Postoperatorio | 60-80 | 30-40 | 40-60 | 40-60 | 1 a 3 |
| 40-60 | 20-30 | 30-50 | 30-50 | 4 a 6 | |
| 30-50 | 15-25 | 20-40 | 20-40 | 7 a 14 | |
| Cirugía menor | |||||
| Preoperatorio | 50-80 | 25-40 | 50-80 | 50-80 | |
| Postoperatorio | 30-80 | 15-40 | 30-80 | 40-60 | 1 a 5, depende tipo del procedimiento |
| Renal | 50 | 25 | 40 | 40 | 3 a 5 |
| Laceración profunda | 50 | 25 | 40 | 40 | 5 a 7 |
| Gastrointestinal | |||||
| Inicial | 80-100 | 40-50 | 60-80 | 60-80 | 1 a 6 |
| Mantenimiento | 50 | 25 | 30 | 30 | 7 a 14 |
| Hemartrosis | 40-60 | 20-30 | 40-60 | 40-60 | 1 a 2 o hasta respuesta |
| Muscular superficial sin afección NV | 40-60 | 20-30 | 40-60 | 40-60 | 2 a 3, o hasta respuesta |
CFC: concentrado de factor de coagulación; SNC: sistema nervioso central; NV: neurovascular; HA: hemofilia A; HB: hemofilia B. Adaptada de Srivastava, et al., 202012 y Guía de Práctica Clínica IMSS, 201717.
Cualquier hemorragia aguda en la PcH debe tratarse a la brevedad posible, de preferencia dentro de las primeras dos horas de haberse producido. Ante la duda sobre la sintomatología en un paciente con hemofilia se tiene que aplicar el CFC. De acuerdo con los lineamientos operativos para la atención del paciente con hemofilia en servicio de urgencias del IMSS,20 toda PcH que ingrese al Servicio de Urgencias por un evento de hemorragia debe catalogarse en triaje rojo brillante.
La aplicación de CFC en bolo IV se calcula considerando el peso ideal de la PcH de la siguiente manera:
FVIII:
Peso del paciente en kg x (% deseado de factor) x (0.5)
FIX:
Peso del paciente en kg x (% deseado de factor)
Debe considerarse la vida media del factor disponible, la pureza, la presencia de otros componentes como el factor de von Willebrand (FvW) o el uso de factor recombinante. El FIX recombinante (FIXr) tiene una respuesta menor que los productos derivados del plasma, de manera que cada unidad de FIX por kilo corporal infundida elevará la actividad del FIX en aproximadamente 0.8 UI/dl en adultos y 0.7 UI/dl en niños menores de 15 años. La razón de la menor respuesta del FIXr no está totalmente establecida.
En caso de no conocerse el tipo de hemofilia se recomienda la administración del concentrado de complejo protrombínico activado (CCPa) a dosis de 50 a 100 U por kg de peso, sin exceder la dosis diaria de 200 U/kg/día.
La respuesta al tratamiento en los casos de hemartrosis aguda se documenta según los criterios de la tabla 9, lo cual permite evaluar la respuesta de forma estandarizada, facilitar la comparación de resultados de diferentes series y tomar decisiones terapéuticas.12
Tabla 9 Criterios para evaluar la respuesta al tratamiento en hemartrosis aguda
| Nivel de respuesta | Respuesta al tratamiento |
|---|---|
| Excelente | Desaparición completa del dolor en 8 h y/o desaparición de los signos de hemorragia después de la primera infusión de factor y sin requerimientos de dosis subsecuentes para el alivio de los síntomas y signos en la misma articulación en 72 h |
| Buena | Mejoría significativa del dolor o de los signos de sangrado en 8 h después de la administración inicial del factor, pero que requiere dosis subsecuentes en las siguientes 72 h para la resolución completa |
| Moderada | Mejoría parcial del dolor o de los signos de sangrado a las 8 h después de la administración inicial del factor y que requiere dosis subsecuentes en las siguientes 72 h, pero sin resolución completa |
| Mala | Nula o mínima mejoría o que empeore el sangrado en las siguientes 8 h de la administración inicial del factor |
Nota: Las definiciones de la respuesta al manejo de una hemartrosis aguda, se refieren al tratamiento con factores de coagulación de vida media estándar y personas con hemofilia con inhibidor negativo. Estas definiciones pueden modificarse para pacientes con inhibidor que reciban agentes puente y en pacientes tratados con factores de coagualción de vida media extendida. Tomada de Srivastava, et al., 202012.
DESMOPRESINA
La desmopresina (DDAVP) es un análogo sintético de la vasopresina que incrementa los niveles séricos de FVIII y del factor von Willebrand, la expresión de FT y estimula la adhesión plaquetaria, por lo que su administración se reserva para pacientes con HA leve.
Dosis:
- 0.3 μg/kg de peso cada 12 h para uso IV o subcutáneo.
- 150 μg en cada narina de spray nasal para adultos > 40 kg de peso.
Debido a la retención de líquidos, la DDAVP debe emplearse con precaución en niños pequeños y está contraindicada en menores de dos años por el riesgo de crisis convulsivas secundario a edema cerebral por la retención hídrica.12
ANTIFIBRINOLÍTICOS
Los agentes antifibrinolíticos como el ácido tranexámico y el ácido épsilon aminocaproico inhiben la fibrinólisis, disminuyendo la activación del plasminógeno en plasmina con incremento de la estabilidad del coágulo. Estos fármacos son útiles en el manejo de hemorragias en mucosas (cavidad oral, nasal y menstrual). Su uso está contraindicado en caso de hematuria, ya que puede impedir la disolución de coágulos en los uréteres, lo que llevaría a una uropatía obstructiva grave.12
ÁCIDO TRANEXÁMICO
En México se cuenta con la presentación en tableta de 650 mg (Lysteda®), la cual se administra de 3 a 4 veces por día vía oral. La presentación inyectable de 500 mg/5 ml (Amchafibrin®) se recomienda 2 a 3 veces por día vía IV. Para pacientes pediátricos la dosis recomendada es 10 mg/kg peso al día por vía IV de 3 a 4 veces por día, o bien 15 mg/kg día en 3 dosis vía oral, con un máximo de 4 g por día.12
ÁCIDO ÉPSILON AMINOCAPROICO
La presentación en México es de solución inyectable en frasco, ámpula de 5 g en 20 ml (Amicar®). La dosis recomendada en infusión IV para adultos es de 4 a 5 g en 250 ml del diluyente administrada durante la primera hora, seguida de una infusión continua de 1 g por hora en 50 ml de diluyente hasta que el cuadro hemorrágico haya sido controlado. La dosis pediátrica es de 100 mg/kg de 3 a 4 veces al día.12
PLASMA FRESCO CONGELADO Y CRIOPRECIPITADOS
Los hemoderivados que contienen factores de coagulación no se someten a procedimientos de inactivación viral ni de priones, por lo que su uso está restringido exclusivamente a casos de urgencia, cuando son la única opción disponible. Se prefiere el uso de crioprecipitados que contienen entre 3 y 5 UI/ml de FVIII, o de forma general 100 UI de FVIII por unidad en lugar de plasma fresco congelado (PFC) para el tratamiento de la HA. El PFC y el plasma desprovisto de crioprecipitado contienen FIX a razón de 1 UI/ml, por lo que pueden utilizarse para el tratamiento de la HB.
Además de las terapias coadyuvantes para el control de sangrado mencionadas previamente, existen medidas de apoyo para el manejo del dolor ocasionado principalmente por la afección musculoesquelética en la PcH y/o durante intervenciones quirúrgicas.12
ANALGESIA
La PcH puede presentar dolor agudo asociado a eventos de hemorragia, como una hemartrosis, y/o de tipo crónico por la artropatía, para lo cual existen recomendaciones para su manejo en adultos o edad pediátrica (Tabla 10).21
Tabla 10 Manejo farmacológico para dolor agudo o crónico en hemofilia
| Niños | Adultos | ||
|---|---|---|---|
| Dolor agudo | Dolor crónico | Dolor agudo | Dolor crónico |
| a) Sin comorbilidades: | a) Sin comorbilidades: | ||
| 1. Paracetamol | 1. Paracetamol ± terapia adyuvante | 1.Paracetamol ± terapia adyuvante | 1. Paracetamol |
| 2. Paracetamol+opioide débil | 2. Paracetamol + opioide débil | 2.Paracetamol + opioide débil o metamizol o COX-2 | 2. COX-2 o AINE no selectivo ± IBP o paracetamol + opioide débil |
| 3. Opioide en hospitalización | 3. Referir a terapia del dolor | 3. Tramadol u opioide fuerte | 3. Tramadol u opioide fuerte ± no opioide |
| b) Daño hepático leve a moderado: | b) Con daño hepático: | ||
| 1. Monitoreo estrecho con el uso de paracetamol y metamizol | 1. Monitoreo estrecho con el uso de paracetamol y metamizol | ||
| 2.Reducir dosis máxima de acuerdo con las guías de prescripción | 2. Reducir dosis máxima de acuerdo con las guías de prescripción | ||
| 3. COX-2 o AINE no selectivo ± IBP solo en pacientes con hepatopatía crónica leve y monitorizando función renal | |||
| c) Con enfermedad o riesgo cardiovascular: | |||
| 1.Los AINE con precaución | |||
| 2.Utilizar de preferencia un inhibidor de COX-2 (naproxeno o ibuprofeno), considerar uso conjunto de IBP | |||
| 3. Evitar uso prolongado de AINE | |||
IBP: inhibidor de la bomba de protones; AINE: antiinflamatorios no esteroideos; COX-2: ciclooxigenasa 2. Adaptada de Auerswald, et al. 201621.
PREPARACIÓN QUIRÚRGICA DE LA PERSONA CON HEMOFILIA
El incremento en la esperanza de vida de la PcH ha condicionado la presencia de complicaciones crónicas propias de la enfermedad, así como de aquellas relacionadas con la edad. Se recomienda la clasificación del riesgo de sangrado en mayor o menor, de acuerdo con el tipo de cirugía (Tabla 11) y de la planeación coordinada del equipo multidisciplinario de atención, antes, durante y posterior al evento quirúrgico.22,23
Tabla 11 Clasificación del riesgo de sangrado en cirugías para personas con hemofilia
| Cirugía general | Ortopédica | Otro | |
|---|---|---|---|
| Mayor | Procedimientos: | Artrodesis y osteotomía | Dental con 3 piezas dentales o más, o extracción del 3er molar |
| «octomía», «otomía», pseudotumor | Reemplazo de articulación o artroplastia | ||
| Sinovectomía | |||
| Reducción de fractura | |||
| Osteosíntesis | |||
| Artroscopia | |||
| Amputación | |||
| Menor | Colocación o extracción de acceso venoso central | Sinovectomía artroscópica, química o por radioterapia | Dental con menos de 3 piezas dentales Cirugía de catarata |
Adaptada de Solimeno, 201823.
La eficacia de la hemostasia durante un evento quirúrgico se puede evaluar por medio de los criterios publicados por la WFH (Tabla 12).12
Tabla 12 Definiciones de respuesta hemostática en procedimientos quirúrgicos
| Excelente | Sangrado intra y posoperatorio similiar (dentro del 10%) a un paciente no hemofílico |
| −Sin necesidad de dosis extra (no planeada) de FVIII/FIX/agente puente y | |
| − Requerimiento transfusional de componentes sanguíneos similar a un paciente no hemofílico | |
| Buena | Sangrado intra y/o posoperatorio levemente mayor de lo esperado para un paciente no hemofílico (entre 10 y 25%), pero la determinación si la diferencia es clínicamente insignificante es valorada por el cirujano/anestesiólogo |
| − Sin necesidad de dosis extra (no planeada) de FVIII/FIX/agente puente y | |
| − Requerimiento transfusional de componentes sanguíneos similar a un paciente no hemofílico | |
| Falla | Sangrado intra y/o posoperatorio mayor de los esperado (25-50%) para un paciente no hemofílico, con necesidad de tratamiento adicional |
| − Necesidad de dosis extra (no planeada) de FVIII/FIX/agente puente o | |
| − Incremento de componentes sanguíneos (dentro de 2 veces) del requerimiento transfusional anticipado | |
| Pobre/Nula | Sangrado intra y/o posoperatorio significativo, que es sutancialmente mayor de los esperado (>50%) para un paciente no hemofílico, que requiere intervención y que no es explicado por causas quirúrgicas/médicas otras que la hemofilia |
| − Hipotensión o traslado inesperado a unidad de cuidados intensivos debido al sangrado o | |
| − Sustancial incremento de componentes sanguíneos (>2 veces) del requerimiento transfusional anticipado |
Notas: Además de las pérdidas sanguíneas estimadas durante la cirugia, los datos de nivel de hemoglobina pre y posoperatorio, y el número de unidades de concentrado eritrocitario transfundidas también son útiles, si son relevantes, para estimar la pérdida quirúrgica de sangre. La evaluación de la hemostasia quirúrgica debe estar a cargo del cirujano y/o anestesiólogo que intervinieron en el procedimiento y completar los registros dentro de las 72 hrs. pos-cirugía. Los procedimientos quirúrgicos se pueden clasificar como menor o mayor, este último se define como aquel que requiere soporte hemostático por un período mayor de 5 dias consecutivos. FVIII: factor VIII; FIX: factor IX. Tomado de Srivastava, et al. 202012.
TRATAMIENTO PROFILÁCTICO
El tratamiento profiláctico consiste en la administración regular del CFC en ausencia de sangrado. Es el tratamiento ideal para pacientes con fenotipo hemorrágico severo, su principal objetivo es evitar o minimizar los eventos de hemartrosis espontáneos y las secuelas por artropatía hemofílica.24-28 En la tabla 13 se describen los tipos de tratamiento profiláctico, los cuales consideran factores como la edad de inicio, edad actual, acceso venoso, fenotipo hemorrágico, estado articular, actividad física, disponibilidad, acceso y presentación del factor para tomar la decisión, en conjunto con paciente y familiar, de la opción del tratamiento más adecuado.12,29-34
Tabla 13 Esquemas de profilaxis en hemofilia
| Tipo de profilaxis | Definición | Objetivo clínico | Evidencia |
|---|---|---|---|
| Profilaxis continua | |||
| Primaria | Aplicación continua* del CFC después del primer evento de hemartrosis en articulaciones grandes†, sin artropatía detectada por clínica o imagen y antes de los tres años | Prevenir o minimizar la incidencia de sangrados y artropatía hemofílica, así como su impacto en el desarrollo psicosocial de los niños y calidad de vida. Permitir actividad física | Ensayos clínicos controlados (JOS24, ESPRIT30) Estudio prospectivo (Canadá) Cohortes retrospectivo (Suecia, Holanda, Alemania, EE.UU., Reino Unido) y comparativos (Francia vs. Holanda y Suecia; Noruega vs. Suecia) |
| Secundaria | Aplicación continua* del CFC después de 2 o más hemartrosis en articulaciones grandes† , pero antes de artropatía crónica documentada por clínica y/o imagen | Reducir el riesgo y frecuencia de sangrados, el desarrollo de articulación diana y/o artropatía; mantener nivel de calidad de vida adecuado. Permitir actividad física | Ensayo clínico controlado (ESPRIT30) Prospectivo controlado (estudio ortopédico de pronóstico) Cohortes retrospectivos (Suecia, Holanda, Alemania, EE.UU., Reino Unido) y comparativo (Francia vs. Holanda y Suecia) |
| Terciaria | Aplicación continua* del CFC después de una artropatía crónica documentada por clínica y radiografías simples | Reducir la frecuencia de sangrado y detener o retrasar la progresión de la artropatía; mejorar calidad de vida; prevenir riesgo de sangrado por comorbilidades. Control del dolor, permitir fisioterapia y/o cirugías ortopédicas. Mejorar calidad vida y actividad física | Ensayo clínico controlado (SPINART29) Prospectivo a largo plazo(POTTER31) Cohorte retrospectivo (Reino Unido, Italia) |
| Profilaxis intermitente | |||
| Profilaxis intermitente | Tratamiento que se aplica para prevenir hemorragias durante periodos que no excedan 45 semanas al año. Casos de rehabilitación, viajes, entrenamientos | Prevención de hemorragias por periodos cortos de tiempo | |
*Aplicación continua: intención de otorgar tratamiento por 52 semanas al año y recibir por lo menos 45 semanas (85%) a priori.
†Articulaciones grandes: tobillos, rodillas, cadera, hombros y codos.
CFC: concentrado de factor de coagulación; JOS: Joint Outcome Study; ESPRIT: Evaluation Study on Prophylaxis: a Randomized Italian Trial; SPINART: Study of Prophylaxis in Adults Randomized Trial; POTTER: Prophylaxis versus ON-demand Therapy Through Economic Report. Adaptada de Instituto Mexicano del Seguro Social, 201717, Coppola, et al., 201432 y Valentino, 201433.
INICIO DEL ESQUEMA DE PROFILAXIS
La evidencia establece que la profilaxis temprana (< 36 meses de edad) es eficaz en términos de calidad de vida y en la reducción del riesgo de daño articular. El estudio ESPRIT (Evaluation Study on Prophylaxis: a Randomized Italian Trial) utilizó FVIIIr a dosis de 25 UI/kg 3/semana y demostró que los pacientes que iniciaron la profilaxis ≤ 3 años de edad tuvieron una menor incidencia de todos los eventos hemorrágicos y de hemartrosis de 0.35 y 0.12 eventos por paciente por mes, respectivamente, en comparación con los pacientes que iniciaron después de los 3 años de edad (0.62 y 0.25). El impacto en la salud articular fue importante, al documentarse que ninguno de los pacientes que inició profilaxis temprana presentó datos radiológicos de artropatía mediante la escala de Pettersson, en comparación con el 46% que iniciaron después de los 3 años. Incluso en pacientes que recibieron el FVIII a demanda se demostró un menor grado de artropatía (57 vs. 85%) con el inicio temprano de la terapia sustitutiva en menores y mayores de 3 años, respectivamente.30
El estudio de Verma que utilizó dosis profilácticas muy bajas de FVIII (10 UI/kg 2 días/semana) documentó la disminución de eventos hemorrágicos en niños < 3 años con solo cinco sangrados en comparación con 19 en ≥ 3 de años durante el seguimiento del estudio de 11.5 meses, con un total de hemartrosis de 0 vs. 11, lo que representa un índice por mes de 0 y 0.15 en menores y mayores de 3 años, respectivamente.35
Profilaxis individual
No existe un parámetro único para evaluar el comportamiento o «fenotipo hemorrágico» de cada paciente. El índice o tasa de sangrado anual (ABR, por sus siglas en inglés: annualized bleeding rate) representa un parámetro clínico de medición de los eventos hemorrágicos y está reconocido como un indicador de buena respuesta al tratamiento profiláctico,36 combinado con estudios de farmacocinética del factor de coagulación.37,38 El reto consiste en identificar a los pacientes que se pueden beneficiar con esquemas de profilaxis con dosis bajas del CFC, sin comprometer el bienestar articular y la calidad de vida. Los esquemas de tratamiento profiláctico se dividen en dos: 1) esquemas con dosis establecidas de CFC (dosis altas, intermedias, bajas o escalonadas), y 2) los esquemas adaptados a las necesidades del paciente.37 En la tabla 14 se mencionan los diversos esquemas de profilaxis primaria con sus ventajas y desventajas.27,36,40
Tabla 14 Esquemas de profilaxis primaria
| Esquema | Dosis de CFC | Ventajas | Desventajas |
|---|---|---|---|
| Sueco (Malmö) | FVIII: 25-40 UI/kg/48 h | Asegura nivel de CFC ≥ 1% Reduce TSAA ~1 | Alto costo Poca adherencia |
| Dosis altas | FIX: 25-40 UI/kg/2 sem | Ideal para pacientes con actividad física | Necesidad de acceso venoso central |
| Holandés (Utrecht) | FVIII: 15-25 UI/kg/2-3 sem | Reduce TSAA 1-2 | Subtratamiento en algunos pacientes |
| Dosis intermedias | FIX: 30-50 UI/kg/1-2 sem | Menor costo que dosis altas Adecuado para adultos | Resultados discretamente peores en SME |
| Canadiense Dosis escalonadas* | FVIII: 50 UI/kg/1 sem 30 UI/kg/2 sem 25 UI/kg/48 h |
Permite entrenamiento en infusión Costo intermedio |
Depende de eventos hemorrágicos para ajuste de
dosis Efecto a largo plazo en SME |
| Dosis bajas | 10-15 UI/kg 1-2 sem | Menor costo Mayor cobertura del número de PcH |
Efecto a largo plazo en SME desconocido, probablemente peor que otros esquemas |
TSAA: tasa de sangrado articular anual; SME: sistema musculoesquelético; PcH: personas con hemofilia; FVIII: factor VIII; FIX: factor IX.
*Indicación para escalonar dosis: ≥ 3 hemartrosis clínicas, o ≥ 4 sangrados significativos en tejidos blandos y/o hemartrosis en un periodo consecutivo de 3 meses, o ≥ 5 hemartrosis en cualquier articulación.
Adaptada de Carcao, et al., 201627 y Berntorp, et al.,201239.
La segunda opción de tratamiento reconoce las múltiples diferencias entre las PcH y ofrece la aplicación de CFC adaptada a las necesidades individuales,28 considerando las siguientes características:
- Fenotipo hemorrágico: considera la heterogeneidad de las PcH (no solo por estudios de farmacocinética) que contribuyen al sangrado y al estado funcional del sistema musculoesquelético; adecúa la terapia de acuerdo con las diversas etapas de la vida (p. ej., disminución de dosis profiláctica en edad adulta por descenso de actividad física). El inconveniente es que se basa en la presencia de sangrados para definir un fenotipo y establecer la profilaxis.
- Actividad física: reconoce la necesidad de una profilaxis intensa en aquellas PcH con altos niveles de actividad física, sin embargo, no toma en cuenta otras variables que pueden favorecer los sangrados y el efecto en el sistema musculoesquelético. El cambio del patrón de actividad física dificulta el ajuste de dosis profiláctica.
- Farmacocinética: calcula de manera científica el régimen profiláctico del paciente para obtener niveles valle en un parámetro determinado y sin necesidad de establecer una profilaxis de acuerdo con el fenotipo de sangrados; tiene el inconveniente de ameritar estudios de farmacocinética (múltiples punciones) y de la pericia del médico para interpretar los resultados; puede ocasionar un tratamiento supraterapéutico o subterapéutico conforme los pacientes pueden requerir diferentes niveles valle para una profilaxis exitosa y no toma en cuenta el patrón de actividad del paciente.
Recomendación: se recomienda que todo paciente con el diagnóstico de hemofilia en la infancia inicie la profilaxis primaria con aplicación del factor de coagulación derivado plasmático en aquellos que nunca han estado expuestos a algún CFC de 2 a 3 veces por semana, dependiendo de la accesibilidad del producto.
CAMBIOS EN ESTILO DE VIDA Y SITUACIONES ESPECIALES
Actividad física
Se debe fomentar el ejercicio para mantener una condición física adecuada y estabilidad osteomuscular, con énfasis en el fortalecimiento de músculos, coordinación y mantenimiento de un peso saludable, lo que impacta en mejorar la autoestima y la calidad de vida. Se recomienda la práctica de deportes o actividades que no impliquen contacto físico como son: caminata, natación, golf, bádminton, arquería, ciclismo, remo, navegación y tenis de mesa, con la administración profiláctica de CFC previo a la actividad. Se sugiere evitar deportes de alto contacto y de impacto como el fútbol, el hockey, el rugby, el boxeo y la lucha, así como actividades de exposición a alta velocidad como carreras de motocross y el esquí.
Previo al inicio de alguna actividad física, ya sea de carácter deportivo o recreacional, la PcH deberá contar con la evaluación de un especialista musculoesquelético o rehabilitador con experiencia en hemofilia, con el objetivo de evaluar si la actividad física es apropiada, recomendar el equipo de protección necesario, el acondicionamiento físico que se requiere previo al inicio de la actividad y, en conjunto con el hematólogo, la planeación de la administración profiláctica de CFC. Los pacientes con disfunciones musculoesqueléticas deberán realizar ejercicios con peso que promuevan el desarrollo y mantenimiento de una densidad ósea adecuada.
Inmunizaciones
Las PcH deben recibir el esquema de vacunación de acuerdo con la edad y el sistema de salud de cada país. La WFH sugiere, en lo posible, la administración subcutánea; en caso de administración intramuscular se recomienda:
- Aplicar posterior a una dosis profiláctica del CFC.
- Colocar una compresa con hielo durante 5 minutos en el sitio de inyección antes de la aplicación.
- Utilizar aguja del menor calibre posible (25-27 G), y
- Compresión por 5 minutos posterior a la aplicación.
Las vacunas con virus vivos atenuados como la vacuna oral de polio o la triple viral pueden estar contraindicadas en PcH e infección por virus de la inmunodeficiencia humana (VIH), pero deben recibir cada año la vacuna contra neumococo e influenza. La inmunización contra hepatitis A y B es conveniente en PcH y de utilidad limitada en personas con infección por VIH. Se había señalado previamente a la vacunación como una “señal de alerta” para el desarrollo de inhibidores, sin embargo, se ha demostrado que no incrementa el riesgo de inhibidores.41
Medicamentos
Debe evitarse el uso de aquellos fármacos que afecten la función plaquetaria, como el ácido acetilsalicílico (AAS) y los antiinflamatorios no esteroideos (AINE), excepto ciertos inhibidores de la ciclooxigenasa 2 (COX-2). El uso de analgésico como el paracetamol (acetaminofeno) es una alternativa segura (Tabla 10).21
Cuidados dentales
Los pacientes deben ser educados para una correcta higiene bucal, con cepillado tres veces al día con cepillo de cerdas de dureza media, uso de pasta dental con flúor de acuerdo con la edad, así como hilo o cepillo interdental para prevenir la formación de caries y enfermedad periodontal. Algunos procedimientos no requieren profilaxis con CFC, como el examen dental, la aplicación de selladores de fisuras y/o pequeñas restauraciones sin anestesia.
Para procedimientos invasivos que requieren anestesia local con infiltración intrapapilar e intraligamentaria, el hematólogo debe indicar la aplicación de CFC de manera profiláctica, DDAVP en caso de hemofilia leve y/o el uso de antifibrinolítico local. Se recomienda que estos procedimientos se realicen en un centro de tratamiento de hemofilia.
Vida escolar
El médico hematólogo deberá emitir las recomendaciones y cuidados generales de la PcH a nivel escolar (de preferencia mediante un documento) a los padres, y estos a su vez deben informar al personal escolar y compañeros de clases, a fin de prevenir actividades con alto riesgo de hemorragias y garantizar la atención inmediata en caso de sangrado o traumatismo.
Adolescencia
Este grupo etario requiere mayor intervención y atención por parte del equipo multidisciplinario de atención de la PcH, en particular de la evaluación psicológica por riesgo de desapego al tratamiento, depresión y dificultades de adaptación, así como de los cambios propios de la edad, como es la actividad física y sexual, entre otros.
Vida sexual
Las potenciales complicaciones hemorrágicas asociadas a la actividad sexual en la PcH están relacionadas con el grado de artropatía y de la limitación al movimiento, como la hemorragia del músculo iliopsoas, por lo que se recomiendan movimientos gentiles y/o maniobras alternas de placer. Semejante a la población general, la PcH puede padecer disfunción sexual, por lo que se pueden valorar las dosis bajas de los inhibidores de la 5-fosfodiesterasa de vida media corta (sildenafilo o vardenafilo) ante la posibilidad de epistaxis por congestión nasal e inhibición de la agregación plaquetaria in vitro. Las infecciones virales por el VIH y/o el virus de la hepatitis C (VHC) tienen un impacto negativo en la actividad sexual por el temor de transmisión a la pareja, por la propia enfermedad o por los efectos secundarios del tratamiento (reducción de la libido o disfunción eréctil).42
Hemofilia en el paciente geriátrico
La esperanza de vida de las PcH se ha incrementado significativamente durante las últimas décadas debido a los avances en el tratamiento y profilaxis, representando un reto para el médico tratante al presentarse las comorbilidades propias de edad avanzada, las cuales se describen de manera general a continuación:43
- Hipertensión arterial sistémica. Algunos estudios observacionales han mostrado que la presión arterial media en PcH es superior a sus controles de población general, por lo tanto, su riesgo de padecer HAS es mayor. De igual manera, se ha observado que requieren de una mayor cantidad de antihipertensivos para mantener cifras de tensión arterial normales.
- Diabetes mellitus (DM). En una cohorte de 294 PcH en Canadá se reportó una prevalencia de DM del 9.9%, sin embargo, no se tienen cifras a mayor escala. La PcH adulta requiere de control y monitoreo metabólico, al igual que la población general, y en caso de DM confirmada se deberán utilizar las mismas guías aplicables a la población local. En caso de aplicación subcutánea de insulina no existe un riesgo de sangrado significativo.
- Obesidad. En las PcH con secuelas por artropatía hemofílica la actividad física está limitada, lo que incrementa en la mayoría de los casos el índice de obesidad. En este grupo de pacientes es necesaria la intervención de fisioterapeutas especializados para aumento de la actividad física e incluso del apoyo de nutriólogos. En las PcH sin complicaciones articulares o musculares se recomienda matener un adecuado índice de masa corporal a través de actividad física de bajo impacto,44 aunado a la profilaxis.
- Enfermedades cardiovasculares. Se ha observado un aumento de enfermedades vasculares trombóticas en pacientes hemofílicos, teóricamente relacionado al uso de CFC, sin embargo algunas publicaciones señalan que la mayoría de los eventos cardiovasculares se presentan en PcH leve o moderada, sugiriendo una aparente protección en hemofilia severa, lo que hasta el momento es controvertido. Los factores de riesgo para ateroesclerosis son los mismos que los de la población general (hiperlipidemia, tabaquismo, síndrome metabólico, hipertensión y enfermedad renal crónica). Las recomendaciones generales para el manejo del síndrome coronario agudo (SICA) es el reemplazo con el CFC, de preferencia mediante infusión continua, manteniendo niveles entre el 30 y el 60% para poder utilizar anticoagulantes y/o antiagregantes plaquetarios. En caso de procedimientos invasivos como cateterismo se recomienda el acceso a través de arteria radial, manteniendo los niveles de actividad del CFC al 80% por lo menos 48 h posteriores al procedimiento. Se pueden utilizar dosis bajas de AAS (100 mg) o doble antiagregante plaquetario como profilaxis antitrombótica posterior a un SICA, generalmente por dos semanas y manteniendo niveles valle del CFC en el 30%.
Parto en mujeres portadoras de hemofilia
Las portadoras con niveles bajos de factor de coagulación deberán considerarse como PcH y recibir el tratamiento adecuado según la gravedad. Durante el embarazo existe un incremento fisiológico del FVIII, pero sin modificarse los niveles de FIX, por lo que se recomienda medir la actividad de los factores durante el tercer trimestre del embarazo con el objetivo de planear una terapia sustitutiva si los niveles están por debajo de las 50 UI/dl. La resolución del embarazo con fetos normales se realizará de acuerdo con las condiciones obstétricas. En caso de sospecha o confirmación de un producto con hemofilia se debe de minimizar el riesgo de traumatismo evitando el uso de fórceps o ventosas durante el parto vaginal, así como los procedimientos invasivos para el feto, como la extracción de sangre y/o la colocación de electrodos internos en cuero cabelludo.
Complicaciones
INHIBIDORES
La complicación más grave relacionada con la aplicación del CFC es el desarrollo de inhibidores contra el factor deficiente. Los inhibidores son aloanticuerpos policlonales que neutralizan la actividad del FVIII o FIX infundido. Son IgG, en su mayoría del subtipo IgG 4, que no fijan complemento y están dirigidas principalmente hacia sitios antigénicos en los dominios A2 y/o C2 del FVIII para el caso de HA.45
Los inhibidores son cuantificados en UB, las cuales se definen como la capacidad del plasma del paciente para neutralizar el factor de coagulación en un pool de plasma normal. Se clasifican en títulos de alta y baja respuesta, con niveles ≥ 5 UB y entre el punto de corte de 0.6 y 5 UB, respectivamente. La detección de un inhibidor clínicamente significativo debe realizarse mediante la técnica modificada de Bethesda-Nijmegen y documentarse en dos ocasiones con un intervalo de 1 a 4 semanas, con un nivel ≥ 0.6 UB y asociado con una recuperación del factor < 66% en una muestra de sangre obtenida 10 a 15 minutos después de haber administrado el factor. Para evitar resultados falsos negativos y detectar inhibidores de baja respuesta se recomienda un periodo de «lavado», que consiste en permitir un tiempo de 48 o 72 h para el FVIII o FIX, respectivamente, entre la última infusión del CFC y la toma de la muestra. En caso de no ser posible, se sugiere un «precalentamiento» de la muestra plasmática del paciente para eliminar el factor residual. Los inhibidores de baja respuesta que desaparecen o disminuyen por debajo del umbral significativo en un periodo de seis meses a partir de su detección y a pesar de la exposición continua del CFC se denominan transitorios y habitualmente no modifican el fenotipo hemorrágico de la PcH.9,12
La incidencia acumulada de inhibidores en HA severa es de alrededor del 30%. En pacientes sin exposición previa al factor de coagulación (PUP, por sus siglas en inglés) el 79% de los casos ocurre dentro de los primeros 20 DE y el restante 21% durante los primeros 75 DE,12 con una diferencia significativa entre los concentrados de factor de coagulación recombinantes (CFCr) y los derivados plasmáticos que se comentará más delante; para el caso de HB severa la incidencia acumulada es del 4-5%, con una media de aparición entre los 9 a 11 DE y sin diferencia entre los productos recombinantes y derivados plasmáticos. Las manifestaciones clínicas de la PcH con inhibidores son una respuesta subóptima para el control de la hemorragia, de acuerdo con los criterios clínicos o con lo esperado por respuestas previas al factor en el mismo paciente, con una mayor morbilidad y complicaciones musculoesqueléticas, por lo que se debe sospechar de la presencia de inhibidor en caso de episodio(s) hemorrágico(s) refractario(s) al CFC en una dosis adecuada, particularmente en pacientes con hemofilia grave.
RIESGO DE INHIBIDORES
El desarrollo de inhibidores contra el FVIII es un mecanismo complejo de interacción entre factores de riesgo genéticos (no modificables) y no genéticos o ambientales (modificables),46-48 los cuales se mencionan en la tabla 15.
Tabla 15 Factores de riesgo para el desarrollo de inhibidores
| Relacionados con el paciente (genéticos y no modificables) | Relacionados con el tratamiento(ambientales y modificables) |
|---|---|
| Tipo de mutación en el FVIII | Número de días exposición |
| Etnia | Edad de la primera exposición |
| Polimorfismos en los genes que modifican la respuesta inmunitaria (IL-10, TNF-a, CTLA4) | Tipo de concentrado de factor de coagulación |
| Historia familiar | Infección intercurrente/estado inflamatorio |
| Severidad de la hemofilia | Exposición intensiva al FVIII |
FVIII: factor VIII; IL-10: interleucina 10; TNF-a: factor de necrosis tumoral alfa; CTLA4: cytotoxic T-lymphocyte antigen 4. adaptada de Coppola, et al.46y Kempton, et al.48
FACTORES GENÉTICOS
De los diversos factores de riesgo genéticos para el desarrollo de inhibidores en HA, el más contundente es el tipo de mutación genética. Las mutaciones que previenen por completo la síntesis del FVIII tienen un mayor riesgo de desarrollar inhibidor (prevalencia > 30%), como son las grandes deleciones y las mutaciones sin sentido (nonsense) con una razón de momios (OR) de 3.6 y 1.4, respectivamente. En contraste, las mutaciones que producen un FVIII disfuncional sin pérdida total de su síntesis tienen un riesgo menor: alteraciones del empalme (splicing) con un OR de 0.95; para las deleciones/inserciones pequeñas y mutaciones con sentido equivocado (missense) el riesgo es de 0.5 y 0.3, respectivamente, con una prevalencia de inhibidor < 10%. Considerando estos aspectos, la revisión de Garagiola clasifica las mutaciones genéticas del FVIII en riesgo alto, intermedio y bajo para el desarrollo de inhibidores49 (Tabla 16).
Tabla 16 Clasificación de las mutaciones del gen del F8 asociadas con el riesgo de inhibidor
| Alto riesgo | Riesgo intermedio | Riesgo bajo |
|---|---|---|
| − Grandes inserciones/deleciones (múltiples exones) | −Grandes inserciones/deleciones (un solo exón) | − Pequeñas inserciones/deleciones |
| −Mutaciones sin sentido en cadena ligera | −Mutaciones sin sentido en cadena pesada | −Mutaciones con sentido equivocado |
| −Inversión del intrón 22 | ||
| −Inversión del intrón 1 | − Mutaciones en sitios de empalme |
Tomada de Garagiola, et al., 201849.
Otros factores genéticos implican la ascendencia, el perfil genético de la respuesta inmunitaria y el antecedente familiar. Miller, et al.7 reportaron una mayor frecuencia de inhibidores de dos veces en PcH A de ascendencia africana e hispanos comparada con los caucásicos (37.1, 46.9 y 19.6%, respectivamente), sin encontrar diferencia entre el perfil de mutaciones en estas poblaciones. Como el espectro de mutaciones en el gen del FVIII no difiere entre etnias, la diferencia podría estar basada en variantes genéticas específicas de cada población, como las regiones reguladoras de genes de citocinas que pudieran modificar el perfil de la respuesta del sistema inmunitario. Se han reportado polimorfismos en estos genes (interleucina [IL] 1b, IL4, IL10, factor de necrosis tumoral alfa [TNF-a] y cytotoxic T-lymphocyte antigen 4 [CTLA4) que tienen una relación significativa con el desarrollo de inhibidores, sin embargo, la asociación de estos factores genéticos aún es controvertida por la baja reproductibilidad de los estudios, de los diferentes perfiles genéticos de las poblaciones analizadas y del reducido número de casos en estudios de cohortes.49 Por último, el antecedente familiar de desarrollo de inhibidores otorga un riesgo absoluto del 48% para esta complicación.
Para el caso de la HB la baja proporción de inhibidores está relacionada con el tamaño inferior de la molécula del FIX de 461 aminoácidos (AA) en comparación con el FVIII (2,332 AA), de la menor cantidad de exones (ocho) y la alta frecuencia de mutaciones genéticas de bajo riesgo para el desarrollo de inhibidores (mutaciones con sentido perdido, sin sentido y de empalme) que causan la producción de una proteína no funcional, pero inmunológicamente detectable en las PcH tipo B. Las PcH B que desarrollan inhibidor tienen la tendencia a presentar una reacción anafiláctica al FIX50 debido a la rápida difusión del FIX al espacio extravascular (por su menor tamaño) y la necesidad de una mayor cantidad de CFC debido a los mayores requerimientos fisiológicos de FIX de 5 μg/ml en comparación con el FVIII de 0.1 μg/ml.47
Las mutaciones causales de hemofilia están compiladas y son de libre acceso en internet en la página de la EAHAD (European Association for Haemophilia and Allied Disorders) en los sitios http://www.factorviii-db.org/ y http://www.factorix.org/ para hemofilia A y B, respectivamente.
FACTORES AMBIENTALES
Los factores de riesgo ambientales que pueden contribuir al desarrollo de inhibidores son múltiples: tipo de FVIII utilizado (plasmático vs. recombinante), los relacionados con la aplicación del CFC (edad de primera infusión, tipo e intensidad de tratamiento) y la infusión asociada con señales de peligro o alarma. La revisión de Peyvandi y Garagiola sintetiza las conclusiones de las principales publicaciones referentes a los factores de riesgo para el desarrollo de inhibidores en PcH A y señala al tipo de producto de FVIII como el principal factor de riesgo ambiental (Fig. 10).51
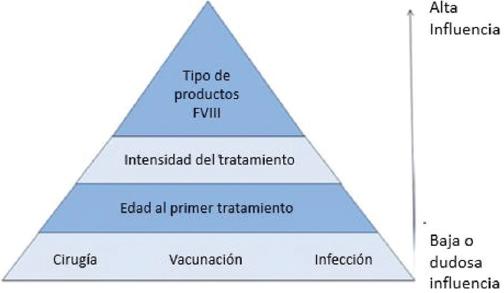
Figura 10 Factores de riesgo ambientales para el desarrollo de inhibidores en hemofilia A severa (tomada de Peyvandi & Garagiola, 201851).
La teoría del modelo de señales de peligro que se publicó en 1994 se basa en el concepto de exposición del factor exógeno en un «ambiente inmunológico de peligro», en el cual el sistema inmunitario está activado por señales de alarma (sangrado severo, cirugía, traumatismo, vacunación e infecciones). La proteína extraña, en este caso el CFC, se presenta de manera intensa (altas dosis y/o tratamiento prolongado) en asociación con señales que activan a los linfocitos T y B, de tal manera que la sola exposición al factor exógeno puede no ser suficiente para el desarrollo de inhibidores.46 Los estudios retrospectiivos en PUP CANAL52 (Concerted Action on Neutralizing Antibodies in severe haemophilia A) y RODIN53 (Research Of Determinants of INhibitor development) documentaron que el tratamiento con dosis altas de FVIII durante cirugías o eventos hemorrágicos incrementaron la tasa de inhibidores y que las dosis profilácticas disminuían el riesgo de inhibidor, sobre todo en los casos de bajo riesgo genético después de los 20 DE e independiente de la dosis empleada.53
PAPEL DE LOS TIPOS DE CONCENTRADOS DE FACTOR DE COAGULACIÓN Y EL DESARROLLO DE INHIBIDORES
La introducción de concentrados de FVIII recombinantes (FVIIIr) a principios de la década de 1990 incrementó el riesgo acumulado de desarrollo de inhibidores en los PUP entre un 25 y un 30%, principalmente en los primeros 50 DE, con un riesgo global de hasta 1.6; IC 95% 1.08 2.37 para el FVIIIr de 2ª generación de cadena completa en comparación con los de tercera generación.54 Los estudios retrospectivos, de revisión y de metaanálisis sugieren una diferencia en la incidencia de desarrollo de inhibidores entre el FVIII derivado del plasma (FVIIIdp) y el FVIIIr,55 pero han sido criticados por sus sesgos metodológicos. En el 2016 se publicó el estudio prospectivo y aleatorizado SIPPET56 (Study on Inhibitors in Plasma-Product Exposed Toddlers) que comparó el desarrollo de inhibidores entre FVIIIdp con FvW y FVIIIr en PUP menores de 6 años con HA severa. Se utilizaron cuatro marcas de FVIIIdp con factor de von Willebrand (Alphanate® y Fanhdi® [FvW:FVIII ~1, Emoclot® y Factane® [FvW:FVIII ~ 0.5) y cuatro productos de FVIIIr (Recombinate®, Kogenate FS®, Advate® y ReFacto AF®o), pero sin la intención de analizar la diferencia entre los diversos productos comerciales. La incidencia acumulada para todo tipo de inhibidores fue del 26.8% para el FVIIIdp y del 44.5% con FVIIr, y para inhibidores de alta respuesta del 18.6 y el 28.4%, respectivamente, documentando una incidencia significativamente mayor del 87% para los FVIIIr. Algunos autores no aceptan los resultados publicados y han criticado aspectos metodológicos del estudio SIPPET, como son: punto de corte de detección de inhibidor, seguimiento menor de 50 DE en un subgrupo de la población, mutaciones de alto riesgo en una alta proporción de pacientes que recibieron FVIIIr, un 50% de pacientes en tratamiento a demanda, mayoría de pacientes provenientes de Egipto, India e Irán y una asignación irregular del grupo de pacientes que recibieron FVIIIr (solo un 16% utilizó de 3.a generación).
Publicaciones posteriores al estudio SIPPET (post hoc) han demostrado que los pacientes tratados con FVIIIr tienen 3-4 veces mayor riesgo de desarrollo de inhibidores en comparación con FVIIdp, sin diferencia significativa mediante modelos de análisis bivariado entre variables de confusión como edad, tipo de mutación, centro de tratamiento, antecedente familiar, exposición previa a hemoderivados y registro de casos censurados (n = 35). El total de los inhibidores se presentó antes de los 39 DE (los de alta respuesta ≤ 34 DE) y el 90% antes de los 20 DE, con un riesgo de desarrollo de inhibidores global y de alta respuesta en los primeros 5 DE de 3.14 (IC 95%: 1.01-9.74) y de 4.19 (IC 95%: 1.18-14.8) respectivamente.57-60
El conocimiento de los factores de riesgo para el desarrollo de inhibidores permite diseñar un esquema de tratamiento individualizado con el objetivo de minimizar esta potencial complicación mediante las siguientes estrategias:
- Asesoramiento genético para los familiares con antecedentes de inhibidores.
- Inicio de profilaxis a edad temprana (habitualmente al terminar el esquema de vacunación al año).
- Evitar extravasación del factor.
- Demorar cirugías electivas.
- Administrar FVIIIdp en las primeras 50 DE.
- En la tabla 17 se señalan las recomendaciones para el monitoreo de inhibidores en la PcH.12
Recomendación:
- Realizar la búsqueda intencionada de inhibidores en PUP o mínimamente tratados a los 20, 50, 100 y 150 DE.
- Realizar la búsqueda intencionada de inhibidores en PTP en caso de cambios en el fenotipo hemorrágico y/o previo a cirugías programadas.
Tabla 17 Recomendaciones para la detección de inhibidores en pacientes con hemofilia
| Después de la exposición inicial a factor, por lo menos cada 6-12 meses, después anual |
| Después de exposición intensa al factor (administración ≥5 dias consecutivos) y dentro de 4 semanas de la última infusión |
| Sangrado recurrente o articulación blanco, a pesar de terapia de reemplazo adecuada con CFC |
| Falla en la respuesta a la terapia de reemplazo adecuada con CFC |
| Recuperación o vida media del factor más baja de lo esperado después de la terapia de reemplazo con CFC |
| Respuesta clínica o de laboratorio subóptima a la terapia de reemplazo con CFC |
| Antes de cirugía |
| Respuesta subóptima a la terapia de reemplazo con CFC en el pos operatorio |
| Pacientes con hemofilia B que presenten una reacción alérgica al FIX incluyendo anafilaxia o síndrome nefrótico |
| CFC: concentrado de factor de coagulación. |
Tomado de Srivastava, et al., 202012.
Tratamiento de inhibidores
El tratamiento de la PcH e inhibidores representa un reto para el médico, principalmente aquellos de alta respuesta, debido a una mayor morbimortalidad asociada a hemorragias y el alto costo de atención. Las estrategias terapéuticas se describen en tres grupos:
Tratamiento de los episodios hemorrágicos a demanda
El tratamiento de los episodios hemorrágicos en PcH A y B con inhibidores dependerá del título de inhibidor (baja o alta respuesta) y de la severidad de la hemorragia. Los pacientes con inhibidores de baja respuesta y hemorragias menores deben recibir dosis mayores de las habituales del CFC (50 a 100/U/kg día) por 2 a 3 días aproximadamente hasta el control del sangrado, monitorizando la respuesta clínica (y/o con niveles del factor deficiente, con el objetivo de mantener concentraciones mayores al 50%). La Fundación Nacional de Hemofilia recomienda calcular la dosis de CFC entre 25-40 UI/kg/UB en casos de inhibidor de baja respuesta, con lo cual se neutraliza el inhibidor y se obtienen niveles hemostáticos para controlar el sangrado. Para hemorragias severas la dosis del CFC deficiente se calculará a 100/U/kg día, con monitoreo clínico y/o con niveles de factor deficiente para garantizar niveles superiores al 50%. En caso de no obtener una respuesta adecuada en pacientes con inhibidor de baja respuesta deberán manejarse como pacientes con inhibidores de alta respuesta.
El manejo ideal de una hemorragia en la PcH con inhibidor de alta respuesta es mediante agentes puente, de los cuales existen dos productos en el mercado: factor VII recombinante activado (rFVIIa) y el complejo coagulante antiinhibidor del FVIII, comúnmente conocido como CCPa. En la tabla 18 se muestran algunas características relevantes de cada componente e información relevante de las fichas técnicas avaladas por la Comisión Federal para la Protección contra Riesgos Sanitarios en México.
Tabla 18 Características de agentes puente
| Denominación genérica (distintiva) | Eptacog alfa activado (NovoSeven) | Complejo coagulante antiinhibidor del FVIII (FEIBA) |
|---|---|---|
| Contenido | FVII recombinante activado | Factores II, IX y X principalmente no activados y FVII activado; concentración máxima de AgFVIII de 0.1 UI/UF* |
| Presentaciones | Liofilizado y diluyente con histidina y agua: 1 mg (1 ml), 2 mg (2 ml) y 5 mg (5 ml) | 500 y 1,000 UF con 20 ml de agua inyectable como diluyente, 80 mg de sodio por vial |
| Almacenamiento y conservación | Conservar por debajo de 30 ºC, protegido de luz. No
congelar Estable química y físicamente después de reconstituir por 6 h a 25 ºC y 24 h a 5 ºC. Utilizarse de inmediato por el riesgo microbiológico |
Conservar entre 2 y 8 ºC. No congelar. Puede conservarse a no más de 25 ºC hasta por 6 meses dentro de la fecha de caducidad. No refrigerar después de almacenar a temperatura ambiente |
| Administración | Bolo de 2 a 5 minutos | Aplicación en 30-45 minutos. Máximo de 2 UF/kg/min |
| Farmacocinética | Vida media efectiva 2 a 3 h. Mayor depuración en pediatría | Vida media efectiva 4 a 7 h |
| Contraindicaciones y precauciones | Hipersensibilidad al producto o a proteínas de ratón, hámster o bovinas. Uso simultáneo con concentrados de complejo de protrombina activados. Potencial desarrollo de trombosis o inducción de CID | Hipersensibilidad al producto Infarto de miocardio, trombosis aguda y/o embolia, CID |
| Antifibrinolíticos | Limitada experiencia con uso concomitante
Factible su uso de manera local en cavidad oral |
Evitar uso concomitante por riesgo de trombosis. Administración en un intervalo de 6 a 12 h después |
| Uso en embarazo y lactancia | Preferible evitar en embarazo, datos limitados
indican que no hay efectos adversos en embarazo o salud
feto/neonato Se desconoce excreción por leche materna, valorar riesgo-beneficio en madre e hijo |
Sin información suficiente para su utilización. Equilibrar riesgos potenciales de eventos tromboembólicos |
| Infusión continua | Reportes con buena respuesta hemostática en pacientes con inhibidor sometidos a cirugía† | Sin estudios que avalen su recomendación |
FVIII: factor VIII; FVII: factor VII; FEIBA: factor eight inhibitor bypassing activity; CID: coagulación intravascular diseminada.
*UF (unidad FEIBA), actividad antiinhibidor del FVIII que acorta el tiempo de tromboplastina parcial activado al 50% en un plasma con alto título de inhibidor.
†Mayor información en la revisión de Holme, et al., 2017.61
Adaptada de A Guide to the Management of Patients with Inhibitors to Factor VIII and Factor IX62.
Ambos productos reportan una eficacia adecuada para el control de eventos hemorrágicos, entre el 80 y el 90%, independientemente del título de inhibidor y con mínimo riesgo de trombosis. El estudio FENOC no encontró diferencia en la eficacia para el control de hemartrosis entre los dos agentes puente,63 sin embargo, existen algunas consideraciones pertinentes en el contexto clínico:
- El FEIBA (factor eight inhibitor bypassing activity) contiene trazas de FIX y FVIII y puede inducir una respuesta anamnésica durante la inducción de tolerancia inmune (ITI), por lo que en estos casos es recomendable el uso de rFVII.
- Al ser el rFVII un producto obtenido mediante tecnología del DNA, se sugiere su empleo en población pediátrica que ha utilizado exclusivamente productos recombinantes.
- La vida media corta del rFVII obliga a infusiones repetitivas.
De la misma manera que en los pacientes con inhibidores de baja respuesta, el manejo de las hemorragias en PcH e inhibidor de alta respuesta dependerá de la severidad del sangrado. En pacientes con sangrado menor se recomienda una dosis única de 270 mg/kg/día de rFVIIa o 90 mg/kg cada 2 h de 1 a 3 dosis; o 50 a 100 UI/kg de FEIBA. En caso de una respuesta inadecuada se deberá tratar como una hemorragia mayor, para lo que se recomienda rFVIIa 90 a 120 ug/kg por dosis cada 2 h o FEIBA 100 U kg/dosis cada 12 h (sin exceder la dosis diaria máxima de 200 U/kg día o de 100 U/kg por dosis), durante las primeras 24 a 48 h considerando una disminución del 50 al 75% del sangrado con una dosis del agente puente y del 90% por lo menos con tres infusiones. Al controlar la hemorragia se puede disminuir la dosis y/o el intervalo de aplicación del agente puente, en caso del rFVIIa cada 3 a 6 h.
En pacientes con inhibidor de alta respuesta y falta de respuesta a un agente puente se sugiere cambiar de fármaco y solo en caso de no obtenerse una respuesta clínica adecuada se puede considerar el uso de combinado de ambos factores de manera secuencial: FEIBA 50 a 100 UI/kg cada 12 horas y posterior a las 4 a 6 h aplicar rFVIIa 90 mg/kg cada 2 h por 2 a 4 dosis.
La evaluación de la respuesta al tratamiento es mediante el monitoreo clínico, en sangrados menores una sola infusión del agente puente puede ser suficiente. Se ha sugerido una correlación entre respuesta clínica y estudios de tromboelastografía y de generación de trombina con la aplicación de rFVII;64 sin embargo, la falta de estandarización dificulta los estudios comparativos. El estudio de Fernández-Bello, et al. analizó la farmacocinética y farmacodinamia del rFVII comparando tres dosis de 90 μg/kg c/3 h y dosis única de 270 μg/kg en hemofílicos sin sangrado, sugiriendo que la dosis alta induce una mayor generación de trombina comparada con la aplicación cada 3 h, sin embargo, esta última mantiene una actividad más prolongada de trombina.65 De tal manera que la decisión de una dosis inicial alta o de cada tres horas dependerá del evento hemorrágico, respuesta clínica y experiencia del equipo médico.
En caso de cirugía en la PcH e inhibidores de alta respuesta se recomienda aplicar previo al procedimiento quirúrgico rFVIIa 90 a 100 μg y continuar su aplicación cada 2 h durante las primeras 24 h, con una eficacia del 90 al 100% o FEIBA 100 U/kg/ cada 12 h.
No se recomienda la administración simultánea de antifibrinolíticos con los agentes puente por el riesgo de trombosis, principalmente en PcH y factores de riesgo protrombóticos.
Profilaxis con agentes puente
El tratamiento profiláctico con agentes puente puede considerarse en pacientes con inhibidores a factores persistentes y fenotipo hemorrágico importante, como son: presencia de una hemorragia grave o potencialmente mortal, tres sangrados significativos en un mismo sitio en un periodo de seis meses o sangrado significativo que requiere de terapia con agente puente más de una vez al mes. El principal objetivo es prevenir o retardar el daño articular. No existe un estudio comparativo de efectividad de profilaxis entre los dos agentes puente, las guías españolas y del Reino Unido recomiendan la profilaxis con rFVII previo a la ITI, otras publicaciones sugieren que ambos agentes pueden usarse de forma profiláctica antes o durante la ITI. La dosis sugerida con rFVIIa es de 90 μg/kg una vez al día o de CCPa de 50 UI/kg en días alternos.
Inducción a la tolerancia inmune
El tratamiento de ITI tiene el objetivo de inducir la tolerancia del sistema inmunitario al factor deficiente contra el cual se desarrolló el anticuerpo neutralizante, reducir los eventos hemorrágicos y permitir la reinstalación del esquema profiláctico. El principio de la tolerancia inmunitaria consiste en la exposición repetida de dosis suprafisiológicas del CFC bajo condiciones no inflamatorias, con o sin el uso concomitante de agentes inmunosupresores.66,67 La primera terapia de ITI para erradicar el inhibidor se realizó en 1974 en la universidad de Bonn (Alemania), utilizando dosis altas de FVIII de 100 UI/kg más CCPa 2/día más tratamiento inmunosupresor.68 Hasta el momento se han descrito varios esquemas de ITI con diferentes dosis de FVIII con tasas internacionales de respuesta del 100% para los casos de títulos de inhibidor de baja respuesta (> 0.6-5 UB/ml) y entre el 60 y el 90% para los de alta respuesta (> 5 UB/ml). Desde hace más de una década se han descrito los factores pronósticos de respuesta para ITI (Tabla 19), sin embargo, una publicación reciente integrada por el grupo de expertos para el manejo de hemofilia A con inhibidores, grupo FIT (The Future of Immunotolerance Treatment)69 reconoce como predictores independientes de respuesta a los títulos de inhibidor antes de la ITI y describe cuatro grupos de riesgo: a) muy buen pronóstico < 25 UB/ml; b) buen pronóstico 25-199 UB/ml; c) mal pronóstico 200-999 UB/ml, y d) muy mal pronóstico > 1,000 UB/ml, y proponen nuevas líneas de tratamiento dependiendo del incremento del título de inhibidor durante el tratamiento, lo cual excede el alcance del presente documento.
Tabla 19 Factores de riesgo para respuesta de ITI
| Características de buen pronóstico | Características de mal pronóstico | |
|---|---|---|
| Edad al inicio de ITI | < 8 años | > 8 años |
| Pico histórico de inhibidor | < 200 UB/ml | > 200 UB/ml |
| Título de inhibidor pre-ITI | < 10 UB/ml | > 10 UB/ml |
| Tiempo para disminución de inhibidor <10 UB antes de iniciar ITI | < 24 meses | > 24 meses |
UB: unidades Bethesda; ITI: inducción de tolerancia inmunitaria. Adaptada de Brackmann,64 et al., 2018.70
La respuesta exitosa de la ITI consiste en negativizar los títulos del inhibidor (< 0.6 UB/ml), recuperación > 66% y una vida media ≥ 6 h del FVIII; una respuesta parcial implica títulos de inhibidor < 5 UB ml, con una recuperación y vida media del FVIII menor del 66% y de 6 h, respectivamente. La falla al tratamiento ocurre con la persistencia del inhibidor > 5 UB/ml.70,71 El estudio aleatorizado de Hay y DiMichele en pacientes con HA grave de «buen pronóstico» con inhibidores de alta respuesta comparó dosis altas de FVII (200 UI/kg/d) vs. dosis bajas (50 UI/kg/3 veces por semana), sin demostrar diferencia significativa en la tasa de éxito (70% en el análisis por intención de tratar). Sin embargo, el tiempo para lograr un título negativo, es decir, la fase con hemorragias más frecuentes, fue significativamente más corto con el régimen con dosis altas.72 En pacientes con pronóstico desfavorable se ha sugerido una mayor tasa de eficacia para inducir tolerancia cuando se administra productos con FVIII que contienen von Willebrand en comparación con productos más altamente purificados, sin embargo no existe hasta el momento evidencia contundente mediante ensayos clínicos controlados.
Infecciones
La transmisión de VIH, VHC y virus de la hepatitis B (VHB) entre 1980 y 1990 mediante los CFC incrementó la morbilidad y mortalidad de las PcH. A partir de la implementación de mejores técnicas para la detección de enfermedades transmisibles por sangre (ETS) y de los procesos de inactivación viral el riesgo de contagio ha disminuido, sin embargo se estima una frecuencia del 30 al 50% de población infectada en países desarrollados. Además, se calcula que la coinfección con VIH y VHC en la PcH incrementa un 59% los costos de tratamiento.73
Los métodos de detección de ETS se han enfocado en retrovirus (VIH y virus linfotrófico humano de células T), VHB y VHC, dejando al descubierto infecciones virales emergentes (virus herpes humano 8, parvovirus B19, hepatitis A y E). Recientemente han tomado relevancia los arbovirus, como el virus del oeste del Nilo, dengue y Chikunguña, y los priones (Creutzfeldt-Jakob), para los cuales no existen protocolos de detección universales en los bancos de sangre. El riesgo de ETS es latente, ya que el 75% de la población mundial no tiene acceso a los CFCr o de derivados plasmáticos que cumplan con los procesos de bioseguridad.
Según la Annual Survey del 2018, en México 45 PcH se encuentran infectadas con VIH y 275 por VHC, sin conocer la cifra exacta de hepatitis activa.4
Complicaciones musculoesqueléticas en hemofilia
La artropatía hemofílica es una variante de enfermedad articular secundaria al sangrado articular (hemartrosis) de repetición y caracterizada por deformidad, hipertrofia sinovial y destrucción de cartílago y hueso. El desarrollo de la artropatía hemofílica ocurre en tres estadios: hemartrosis aguda, sinovitis crónica y artritis degenerativa.
FISIOPATOLOGÍA
La fisiopatología de la artropatía hemofílica es diferente a la de procesos inflamatorios sistémicos o degenerativos como la artritis reumatoide u osteoartritis degenerativa, respectivamente. Su principal mecanismo está desencadenado por la liberación de hemoglobina (Hb) y depósito de hemosiderina en el tejido sinovial, lo que induce una hipertrofia de la membrana sinovial y una neovascularización. Ambos procesos son mediados por citocinas inflamatorias como la IL-6, la IL-1b, el TNF-α, el interferón gamma y la IL-8, que ocasionan una sinovitis crónica con la formación de vellosidades hacia el interior del espacio articular, las cuales son más susceptibles a hemorragias espontáneas o al mínimo estrés, lo que establece un círculo vicioso de resangrado y mayor sinovitis. Por otra parte, los cambios metabólicos (activación de metaloproteinasas) en cartílago y hueso subcondral promueven su destrucción, con la subsecuente deformación articular. Para una mayor descripción de la fisiopatología de la artropatía hemofílica se puede revisar el trabajo de Wyesure, et al.74
DIAGNÓSTICO
Las valoraciones de salud articular y cambios estructurales se realizan mediante el examen clínico y estudios de imágenes. Las radiografías simples demuestran los cambios óseos avanzados como crecimiento excesivo de epífisis, estrechamiento del espacio articular, formación de quistes subcondrales y osteoporosis, pero tienen poca sensibilidad para demostrar los cambios tempranos en tejidos blandos que ocurren antes del daño irreversible del cartílago. Los dos sistemas de clasificación radiológica son: la escala de Arnold-Hilgartner, la cual valora los cambios en partes blandas y tejido óseo, y en la que la imagen con mayor nivel de afección dictamina el grado de artropatía (grado I a V); y la escala de Petterson, que excluye los tejidos blandos y asigna una puntuación para cada cambio radiológico. Esta última escala de evaluación es la recomendada por la WFH por su disponibilidad y costo accesible.
Los estudios de radiografía simple solo detectan cambios irreversibles, por lo que la resonancia magnética (RM) se describe como el estándar de oro para el diagnóstico de la artropatía hemofílica al identificar cambios tempranos (reversibles) en tejidos blandos, como: integridad del cartílago, depósitos de hemosiderina, líquido intraarticular y edema. Los inconvenientes de la RM son la necesidad de sedación en niños, el costo, la disponibilidad y la incapacidad para explorar múltiples articulaciones. La alternativa a estos inconvenientes es el ultrasonido (US), el cual, con la tecnología adecuada, permite obtener imágenes de alta resolución de la anatomía articular, incluyendo tendones, ligamentos y músculos, representado una importante herramienta auxiliar para la detección y seguimiento de hemartrosis aguda, así como para el diagnóstico diferencial entre el dolor por sangrado o por afección de partes blandas (esguinces, desgarre muscular, tendinitis, etc.). En los últimos años se ha sugerido el uso de una escala de evaluación mediante US que valora la cápsula sinovial, el cartílago y el hueso subcondral, denominada Haemophilia Early Arthropathy Detection with UltraSound (HEAD-US).
La WFH recomienda la evaluación del deterioro articular de las seis articulaciones índice (codos, rodillas y tobillos) mediante la escala del examen físico de Gilbert, la cual está diseñada para adultos y niños mayores con artropatía ya establecida, sin embargo tiene deficiencias como la falta de fiabilidad, validez y sensibilidad a cambios más pequeños en pacientes con enfermedad menos grave, por lo que se han realizado varias modificaciones al sistema. La segunda escala clínica y funcional de las articulaciones que valora entre otros datos la marcha, arcos de movimiento, tono muscular, dolor y aumento de volumen se realiza mediante el sistema de puntuación de salud articular Hemophilia Joint Health Score (HJHS). Dependiendo del contexto del paciente y la experiencia del equipo multidisciplinario, las escalas de salud articular deben aplicarse para ayudar en el diagnóstico de la sinovitis crónica y guiar las decisiones del tratamiento.12
Nuevos tratamientos
Introducción
La terapia de reemplazo con el CFC ha sido eficaz en el control y/o prevención de sangrado en PcH durante décadas, sin embargo está limitada por la accesibilidad y conservación de los productos, la relativa corta duración hemostática y el desarrollo de complicaciones como la aparición de anticuerpos neutralizantes (inhibidores) contra el FVIII o el FIX. La búsqueda de la curación sigue siendo el objetivo final. Con la intención de mejorar el tratamiento de las PcH se han diseñado productos con vida media más prolongada, y mecanismos de acción alternativos para lograr una decuada hemostasia e inmunogenicidad reducida75 por medio de: a) CFC con vida media extendida (VME); b) terapia génica; c) anticuerpos específicos que simulen la función del FVIII, y d) moléculas que modifican la acción de anticoagulantes naturales, las cuales se muestran y se describen brevemente en la figura 11.76
Figura 11 Mecanismos de acción de las terapias emergentes en hemofilia: 1) CFCr con vida media extendida; 2) terapia génica; 3) anticuerpos que simulen la función del FVIII (emicizumab), y 4) moléculas que modifican la acción de anticoagulantes naturales (anticuerpos anti-TFPI [concizumab, BAY 1093884, BAX 499]), anti-APC, bloqueo de AT (Fitusiran®), etc. (adaptada de Arruda, et al., 201876). CFCr: concentrado de factor de coagulación recombinante; FVIII: factor VIII; AAV: virus adenoasociado; TFPI: inhibidor de la vía del factor tisular; APC: proteína C activada; AT: antitrombina.
Factores de coagulación con vida media extendida
Los CFC de VME tienen la facultad de reducir el número de infusiones necesarias para tratar los episodios de hemorragias agudas y extender el tiempo entre infusiones profilácticas, permitiendo así una mayor adherencia a los esquemas de tratamiento. Las estrategias para retardar el aclaramiento y prolongar la vida media del FVIII o el FIX recombinante en el espacio intravascular es mediante la unión artificial a otras moléculas a través de las siguientes opciones:
- Unión con el fragmento cristalizable (Fc) de las Ig.
- Pegilación.
- Unión a la albúmina recombinante.
FACTOR VIII DE VIDA MEDIA EXTENDIDA
El beneficio de los productos de FVIII de VME ha sido limitado, con un promedio de extensión de la vida media de 1.5 veces, lo que ha permitido la aplicación profiláctica en adultos de dos veces por semana, pero con un amplio rango en la vida media entre pacientes y menor duración en población pediátrica, por lo que se deben personalizar las dosis de acuerdo con el fenotipo de sangrado y las vidas medias del producto estándar y de VME.77
La primera tecnología para incrementar la vida media del FVIII fue mediante la fusión con la región constante (Fc) de la IgG. El efmoroctocog alfa (Elocta®, Biogen/Sobi) es un análogo del factor FVIII unido al dominio Fc de la IgG1 humana sin dominio B. La segunda opción para prolongar la vida media del factor es mediante la unión covalente de polietilenglicol (PEG) al FVIII (pegilación).78 Existen tres productos aprobados por la Food and Drug Administration (FDA) con esta tecnología:
- Octocog alfa® (Adynovate, Shire).
- Turoctocog alfa pegol® (Novoeight-GP, Novo Nordisk).
- Damoctocog alfa pegol® (DAMATO Bayer).
El tercer mecanismo para disminuir el aclaramiento del FVIII es mediante la adición de cargas negativas por medio de ácido polisiálico, que interfiere en la depuración mediada por el receptor. La molécula BAX826 (Octocog alfa®, Shire) está en fase de estudios preclínicos.
FACTOR IX DE VIDA MEDIA EXTENDIDA
El esquema tradicional de profilaxis en la PcH B severa es mediante la infusión de FIX dos veces por semana. Las modificaciones estructurales de los productos de FIX con VME incluyen, al igual que el FVIII, la pegilación y fusión con Fc o albúmina. El primer FIXr de VME en el mercado fue el fusionado a la proteína Fc (FIXr-Fc) (Eftrenonacog alfa®, Alprolix, Biogen/Sobi), con una vida media de 86.5 ± 32.2 h. Los pacientes que recibieron 50 UI/kg semanalmente lograron niveles mínimos de FIX de 1-3 UI/dl, con un descenso rápido en las primeras 24 a 72 h postinfusión, seguido de una vida media más prolongada. El segundo factor aprobado por la FDA es el FIXr unido a albúmina (Albutrepenonacog alfa®, Idelvion, CSL Behring), el cual tiene la ventaja sobre la farmacocinética del FIXr-Fc de un descenso gradual posterior a la infusión, con una vida media de 104 h. La extensión de la vida media de este producto se basa en su alto peso molecular (por encima del umbral renal) y una interacción dependiente del pH con el receptor neonatal de Fc, que evita su degradación intracelular. Por último, el FIXr pegilado nonacog beta pegol (N9-GP), manufacturado por la empresa Novo-Nordisk y aprobado por la FDA en mayo del 2017, bajo el nombre comercial de REBINYN®.
Estrategias de tratamiento sin factor sustitutivo
La principal ventaja de esta modalidad de tratamiento es minimizar el riesgo de desarrollo de inhibidores, la administración subcutánea e intervalos prolongados de aplicación semanal y/o mensual. Estas terapias intentan amplificar la generación de trombina por medio de diferentes mecanismos de acción,79 o bien, incrementar la producción endógena del factor deficiente mediante una terapia génica, los cuales se explican a continuación.
TERAPIAS QUE AMPLIFICAN LA GENERACIÓN DE TROMBINA
Anticuerpo biespecífico que imita la función del factor VIII
El emicizumab, autorizado por la FDA con el nombre comercial de Hemlibra® (Hoffmann La-Roche), es un anticuerpo monoclonal humanizado biespecífico que simula la función biológica del FVIIIa, al establecer un efecto procoagulante mediante sus regiones de unión a antígeno (Fab, por sus siglas en inglés: antigen-binding fragment) entre el FIXa y su sustrato el FX de la coagulación sobre una capa de fosfolípidos, generando trombina con un efecto dependiente de la dosis y, por lo tanto, acortando el TTPa. La administración es subcutánea, con una vida media aproximada de 4-5 semanas. La dosis autorizada para el manejo de la PcH A con inhibidores es de 3 mg/kg semanal las primeras 4 semanas y, posteriormente, 1.5 mg/kg semanal o 3 mg/kg quincenal o 6 mg/kg mensual. No comparte homología estructural con el FVIII, excepto por los sitios de unión, por lo que no se espera el desarrollo de inhibidores contra esta molécula y no es neutralizado por los inhibidores del FVIII.76,80
Los estudios HAVEN 181 y 2 evaluaron PcH A con inhibidores de alta respuesta, reportando una reducción de la tasa anual de hemorragia con una diferencia significativa del 87% con aplicación semanal de emicizumab. Con base en estos estudios, el emicizumab fue aprobado originalmente en 2017 como profilaxis para evitar o reducir la frecuencia de episodios de sangrado en PcH A con inhibidores dirigidos contra el FVIII. Es pertinente mencionar la presencia de complicaciones trombóticas con el uso concomitante de CCPa a dosis terapéuticas secundario a un sinergismo entre las dos sustancias, ya que el emicizumab incrementa 20,000 veces la acción enzimática del FIXa contenido en el CCPa, por lo que se recomienda su empleo, en caso necesario, a dosis bajas. No se reportan eventos trombóticos asociados con el uso de rFVIIa ni con emicizumab como monoterapia.
Posteriormente, en 2018 se extendió la indicación de uso de emicizumab a PcH A sin inhibidores, con base en los estudios clínicos HAVEN 3 y HAVEN 4,82 que demostraron reducción de la tasa anual de sangrados de entre el 96 y el 97% en comparación con placebo, así como una mediana de 0. A la luz de esta indicación, se ha sugerido considerar el uso de emicizumab en pacientes sin inhibidores con acceso venoso difícil, no candidatos a uso de catéter venoso central, que requieran dosis altas de FVIII (con comportamiento clínico similar a pacientes con inhibidores) o con alto riesgo de inhibidores.
Agentes que modifican la función de los anticoagulantes naturales como el inhibidor de la vía del factor tisular, la antitrombina y la proteína C activada
Inhibidor de la vía del factor tisular
En la PcH la amplificación de la coagulación y generación de trombina está alterada por la deficiencia del FVIII o del FIX. El inhibidor de la vía del FT (TFPI, por sus siglas en inglés: tissue factor pathway inhibitor) es una serinproteasa que desempeña una función importante en la generación inicial de trombina mediante la inhibición del complejo factor tisular-factor VIIa y de la protrombinasa. Se encuentran en proceso de investigación anticuerpos monoclonales y aptámeros (ácidos nucleicos de cadena sencilla de ADN o ARN con alta especificidad de unión a una molécula diana [proteína]) que inhiben la función del TFPI: Concizumab® (Novo Nordisk), anticuerpo monoclonal humanizado de tipo IgG4; BAY 1093884® (Bayer), anticuerpo monoclonal humanizado IgG2, y BAX 499® (Shire), aptámero de ácido nucleico pegilado.
Inhibidor de la antitrombina (Fitusiran®)
Fitusiran® (farmacéutica Alnylam) es un ácido ribonucleico de interferencia (ARNi), el cual se une al ARN mensajero (ARNm) e interrumpe su producción, con la subsecuente disminución de la síntesis de antitrombina (AT) a nivel hepático. La AT es el principal anticoagulante natural que inactiva a la trombina y al FXa. La disminución de los niveles de AT en pacientes que han recibido diferentes dosis subcutáneas (SC) de Fitusiran® ha reportado un incremento en la generación de trombina y una disminución en el promedio del sangrado anual. Por motivos de seguridad y eficacia se ha rediseñado el ensayo clínico fase 3 ATLAS con la administración mensual del inhibidor de la antitrombina.79
Terapia génica
La terapia génica consiste en la introducción de la secuencia de un gen específico en una célula blanco. La utilización de un virus como vector del material genético se denomina transducción y se puede realizar mediante dos estrategias: a) administración directa in vivo del gen terapéutico por medio de un vector principalmente asociado a adenovirus (AAV, por sus siglas en inglés: adeno-associated virus), y/o b) trasplante de células a las que se ha insertado el gen ex vivo, mediante vectores de tipo lentivirus.83
La terapia génica en hemofilia utiliza el AAV para transducir el gen del factor de coagulación directamente en los hepatocitos. Algunos ensayos clínicos han obtenido la expresión sostenida de niveles terapéuticos de FVIII y FIX, sin embargo, tiene sus limitantes, ya que alrededor de un 40% de la población tiene anticuerpos contra la cápside de alguno de los serotipos del AAV, lo que limita la transducción, así como el desarrollo de una respuesta inmunitaria de tipo celular caracterizada por una transaminasemia y/o una disminución de la expresión transgenética.
Las investigaciones actuales de terapia génica en hemofilia están enfocadas en la administración de vectores AAV directamente a nivel hepático vía IV, con el control de la respuesta inmunitaria mediante altas dosis de esteroide. Estudios recientes en PcH B sugieren una potencial cura para esta enfermedad.
La segunda parte del presente documento revisa una coagulopatía adquirida por inhibición de la actividad del FVIII, que en los últimos años ha tomado relevancia en México por su impacto en la morbimortalidad, presentación clínica y su alto costo de atención.
Hemofilia A adquirida
La HAA es un padecimiento de origen autoinmune, causado por autoanticuerpos policlonales de subclase IgG1 e IgG4 dirigidos contra epítopes específicos de la cadena pesada (dominio A2) o cadena ligera (dominios C2 y A3) del FVIII que ejercen su efecto inhibitorio interfiriendo en la unión al FIXa, fosfolípidos de membrana y FvW. La incidencia de HAA es de 0.2 a 1.48 casos en 1,000,000 habitantes por año. El promedio de edad de presentación es de 65 años, con una distribución bifásica: un pequeño pico observado en mujeres entre los 20 a 30, generalmente asociado a embarazo o colagenopatías, y el segundo sin predominio de sexo en mayores de 60 años. El rango de mortalidad general es amplio (15-42%) debido a complicaciones asociadas a la edad avanzada de la mayoría de los casos, comorbilidades y/o efectos secundarios al tratamiento.84,85
Se considera que la formación de autoanticuerpos proviene de una pérdida de los mecanismos de tolerancia inmunitaria periférica, resultado de factores genéticos y ambientales. La mayoría de los casos de HAA son idiopáticos (43.6-51.9%), seguidos de alguna neoplasia como tumores sólidos (cáncer de próstata o pulmón), trastornos linfoproliferativos (comúnmente linfoma) entre el 6.4-18.4%, enfermedades autoinmunes como artritis reumatoide (9.4-17.0%), ingesta de medicamentos, enfermedades dermatológicas, embarazo (habitualmente 1 a 4 meses posparto) y otros. Los casos pediátricos de HAA son raros, con una incidencia estimada en menores de 16 años de 0.045 casos por millón de habitantes por año.86
Diagnóstico
La HAA debe sospecharse en aquellos casos que presentan un sangrado de inicio súbito, frecuentemente severo, de manera espontánea o posterior a trauma leve, procedimientos invasivos o posparto, sin historia personal o familiar de hemorragia, un TTPa prolongado que no corrige con la mezcla de plasma normal, pruebas hemostáticas en parámetros normales (conteo de plaquetas, TP, TT y fibrinógeno) y anticoagulanmte lúpico negativo.87 El patrón hemorrágico es diferente al de la hemofilia congénita, la mayoría de los pacientes presentan hematomas o equimosis subcutáneos (> 80%), sangrado muscular (> 40%) o gastrointestinal (> 20%); los sangrados a nivel genitourinario o retroperitoneal, posparto y en otros sitios representan menos del 10%. De manera frecuente las hemorragias ponen en riesgo la vida de los pacientes por la intensidad del sangrado o afección a nivel cerebral. El principal dato por laboratorio para considerar una HAA es la presencia de un TTPa prolongado que no corrige con plasma normal y un TP normal. El diagnóstico diferencial de un TTPa prolongado involucra la presencia de un anticoagulante lúpico (AL) o el efecto de heparina. La incubación de plasma del paciente con plasma normal 1:1 por 2 h a 37 °C permite evidenciar la presencia de un inhibidor al presentarse una mayor prolongación del TTPa por la inactivación dependiente de la temperatura y tiempo del FVIII. La prolongación similar del TTPa antes y después de la incubación es indicativa de un AL. Las heparinas no fraccionadas y de bajo peso molecular (HBPM) a dosis terapéuticas pueden prolongar el TTPa, sin embargo, la heparina convencional también prolonga el TT con un tiempo de reptilasa normal; para descartar el efecto de HBPM se necesita de ensayos anti-factor Xa.
La confirmación de HAA se realiza mediante la documentación de niveles bajos de la actividad del FVIII y evidencia de un inhibidor cuantificado en UB, de preferencia con la técnica de Bethesda-Nijmegen o con el ensayo Bethesda. En algunos centros se realiza la detección de auto anticuerpos IgG anti-VIII por ELISA (enzyme-linked immunosorbent assay).84 La clasificación es semejante a la hemofilia congénita y depende de los niveles de inhibidor en títulos bajos (< 5 UB/ml) o altos (> 5 UB/ml). La figura 12 muestra el algoritmo diagnóstico para hemofilia adquirida sugerido1 por el grupo español.88
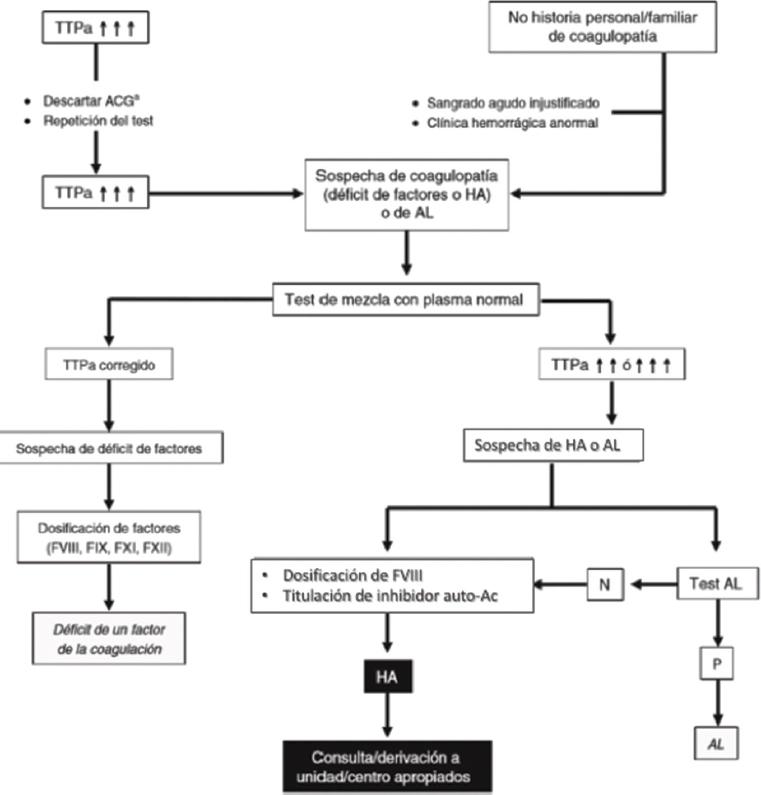
Figura 12 Algoritmo diagnóstico por laboratorio de HAA (tomada de Mingot-Castellano, et al., 201788). aDescartar tratamiento con ACG y/o contaminación con heparina. ACG: anticoagulante; AL: anticoagulante lúpico; auto-Ac: autoanticuerpo; FVIII: factor VIII; FIX: factor IX; FXI: factor XI; FXII: factor XII; HA: hemofilia adquirida; N: negativo; P: positivo; TTPa: tiempo de tromboplastina parcial activada.
Los autoanticuerpos en HAA presentan un patrón de inactivación del FVIII no lineal o tipo 2 (a diferencia de la cinética tipo 1 causada por aloanticuerpos característicos en hemofilia congénita), ocasionando una primera fase de inactivación lineal, seguida de una fase de equilibrio o «meseta» que permite la detección residual del FVIII, lo cual se muestra en la figura 13. Este mecanismo provoca que los niveles de FVIII residual no sean fiables para predecir el riesgo de hemorragia, por lo tanto, aun con niveles relativamente altos de FVIII el riesgo de sangrado es importante si los títulos del inhibidor son altos en HAA.(ídem)
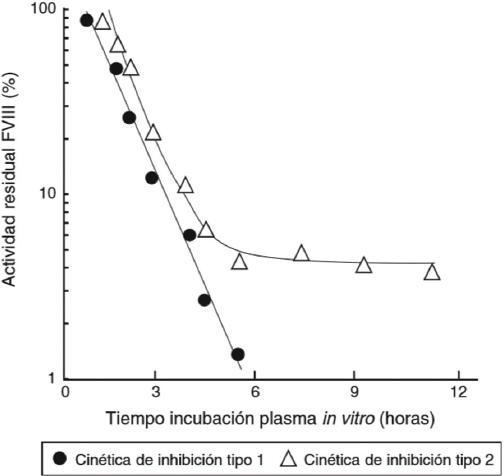
Figura 13 Cinética de inhibición del FVIII por anticuerpos. La cinética de inhibición de la actividad del FVIII tipo 1 (círculos negros), característica de aloanticuerpos en hemofilia congénita, presenta un patrón lineal donde a mayor concentración de anticuerpos mayor es la neutralización del FVIII. La cinética tipo 2 (triángulos) provocada por autoanticuerpos en hemofilia adquirida presenta una fase de inhibición lineal primaria, seguida de una fase de equilibrio, lo que permite detección de actividad residual de FVIII in vitro (adaptada de Mingot-Castellano, et al., 201788). FVIII: factor VIII.
Tratamiento
El 70 a 90% de los casos de HAA presentan una hemorragia mayor, por lo que el manejo del paciente con HAA debe realizarse de manera inmediata por personal con experiencia en esta patología, o de no ser posible, bajo su asesoramiento, en centros que cuenten con los recursos suficientes de factores hemostáticos y para el monitoreo de niveles de inhibidor y FVIII.
Las metas generales del tratamiento en HAA son:
Control y prevención del sangrado
Se recomienda iniciar el tratamiento hemostático (antihemorrágico) en pacientes con HAA con síntomas de sangrado mayor y/o disminución del nivel de Hb, independientemente del título de inhibidor y de los niveles de FVIII, o de manera profiláctica en pacientes con alto riesgo de sangrado (cirugía o parto reciente, úlcera péptica, etc.). Pacientes con sangrado leve o moderado y sin repercusión significativa en el nivel de Hb pudieran no ameritar terapia hemostática inmediata, pero requieren de una vigilancia estrecha. Las medidas preventivas de sangrados involucran el diferimiento de procedimientos invasivos o cirugías hasta la erradicación del inhibidor y documentación de niveles de FVIII adecuados; en caso de intervención urgente se deben utilizar agentes puente antes y después del procedimiento (comentado previamente). Una tercera parte de los casos no presentan sangrado y no ameritan de manejo hemostático. En la tabla 20 se describen las alternativas de tratamiento para hemorragia aguda en HAA.84,85,89 Cualquiera de los dos agentes puente (bypassing) disponibles en el mercado (rFVIIa y FEIBA) son de primera opción de tratamiento para la hemorragia en HAA. El registro europeo EACH2 (European Acquired Haemophilia)90 de 237 pacientes tratados con rFVIIa o FEIBA no reportó diferencia significativa para el control de sangrado, con tasas de respuesta del 92 y el 93%, ni en la incidencia de trombosis (2.9 y 4.8%, respectivamente) Las ventajas y desventajas de los agentes puente se describen en la Tabla 21, las cuales deben valorarse de acuerdo con el contexto clínico del paciente, por ejemplo, en el posparto se recomienda el rFVIIa.85
Tabla 20 Alternativas de tratamiento hemostático en HAA
| Agente | Observaciones |
|---|---|
| Tratamiento de primera línea | |
| Factor rVIIa | 70-90 µg/kg c/2-3 h hasta control de sangrado, después disminuir dosis y/o aumentar intervalo de aplicación |
| CCPa | 50-100 U/kg c/8-12 h, sin exceder 200 U/kg/día |
| Tratamiento alternativo (si la terapia puente no está disponible) FVIIIdp o FVIIIr | |
| Desmopresina | Solo en caso de inhibidor de baja respuesta y/o sangrado menor |
| Escasa evidencia. Considerar en sangrado menor y títulos muy bajos de inhibidor (< 2 UB/ml) y FVIII > 5%. Valorar potenciales efectos secundarios | |
| Antifibrinolíticos | Potencial utilidad en sangrado oral y contraindicado en caso de hematuria |
| Tratamiento alternativo* | |
| FVIIIr porcino | 100-200 U/kg inicial, monitoreo de actividad de FVIII para recalcular dosis |
| Tratamiento de segunda línea
Inmunoabsorción y/o plasmaféresis |
Reservado para hemorragias refractarias o reducción urgente de inhibidor previo a cirugía |
*Disponible solo en Europa, Canadá y EE.UU.
HAA: hemofilia A adquirida; Factor rVIIa: factor VII recombinante activado; CCPa: concentrado de complejo protrombínico activado; FVIII: factor VIII; FVIIdp: factor VIII derivado plasmático; FVIIIr: factor VIII recombinante. Adaptada de Franchini, et al., 201784, y Kruse-Jarres, et al., 201785.
Tabla 21 Ventajas y desventajas de agentes puente
| Agente | Ventaja | Desventajas |
|---|---|---|
| Complejo coagulante antiinhibidor del factor VIII (FEIBA) | Eficacia comprobada para control de sangrado | No existe monitoreo específico por laboratorio
Riesgo de trombosis arterial o venosa |
| Factor VII recombinante activado (rFVIIa) | Eficacia comprobada para control de sangrado | No existe monitoreo específico por laboratorio
Vida media corta (2 h) Riesgo de trombosis arterial o venosa |
Adaptada de Kruse-Jarres, et al., 201785.
Consideraciones relevantes de las terapias hemostáticas en hemofilia A adquirida
La elección del agente puente dependerá de la disponibilidad, aspectos económicos, eficacia previa en caso necesario y experiencia del médico. La respuesta al tratamiento se realiza mediante valoración clínica del paciente habitualmente en las primeras 24 a 72 h dependiendo del sitio, tipo y severidad de la hemorragia. Una vez controlado el sangrado se debe disminuir la dosis y/o el intervalo de administración del agente puente para prevenir el riesgo de trombosis. Zanon, et al. reportaron que el uso de dosis bajas profilácticas de FEIBA (media 54.2 ± 23 UI/kg) por un promedio de 20 ± 17.6 días previno de manera significativa la tasa de resangrado.91
Las terapias alternativas como la DDAVP y los concentrados de FVIII (derivado plasmático o recombinante) tienen el fundamento de aumentar los niveles de FVIII, con una respuesta del 68.3%. La DDAVP incrementa los niveles de FVIII de manera impredecible, lo que representa un factor de riesgo para trombosis en pacientes de edad avanzada y condiciones predisponentes (malignidad o antecedente de trombosis). En pacientes hospitalizados y con factores de riesgo para trombosis se debe considerar la tromboprofilaxis con niveles de FVIII mayores del 50% y evitar los agentes puente en pacientes sin evidencia de sangrado activo o mayor riesgo de hemorragia. Los agentes antifibrinolíticos se pueden utilizar como adyuvantes de manera tópica en forma de colutorios y están contraindicados por un periodo de 12 h después de utilizar FEIBA. Por último, el FVIII recombinante porcino tiene una efectividad del 86%, sin eventos adversos significativos, sin embargo no está disponible en México.
No existe un parámetro de laboratorio validado para el monitoreo de la terapia hemostática, el TTPa no es un buen indicador de respuesta al tratamiento, consolidando la importancia de la evaluación clínica de los pacientes. En la tabla 22 se describen los criterios de falla al tratamiento en HAA.
Tabla 22 Definiciones de falla al tratamiento en HAA
| Sangrado persistente, sin cambios en pérdida de sangre en transcurso de tiempo |
| Nivel de Hb sin cambios o descenso a pesar de terapia transfusional |
| Incremento de dimensiones de sangrado interno por estudios de imagen |
| Evidencia de sangrado continuo después de 48 h de tratamiento adecuado (24 h para sitios anatómicos críticos) |
| Sangrado en nuevos sitios aun con terapia antihemorragia |
| Incremento de dolor asociado a hematomas a pesar del tratamiento |
| HAA: hemofilia A adquirida; Hb: hemoglobina. |
Adaptada de Huth-Kühne, et al., 200989.
Erradicación del inhibidor
Todo paciente adulto con diagnóstico establecido de HAA debe recibir terapia de inmunosupresión (TIS) de manera inmediata, con el objetivo de eliminar la clona celular responsable de la síntesis de autoanticuerpos. En la tabla 23 se describen las opciones y dosis recomendadas de esquemas de TIS de primera línea, las cuales se deben de invidualizar de acuerdo a las condiciones generales del paciente, comorbilidades y factores de riesgo, ya que los casos con FVIII <1% de actividad residual habitualmente requieren mayor tiempo de tratamiento y/o una TIS combinada para obtener respuesta.85
Tabla 23 Opciones para terapia de inmunosupresión (TIS) de primera línea en HAA
| Primera línea recomendada | Dosis recomendada | Comentarios |
|---|---|---|
| Monoterapia con corticosteroides | Prednisona 1 mg/kg VO diario (alternativa dexametasona 40 mg VO o IV diario x 4-7 días)* | Probablemente no efectivo en ≤3 semanas en
pacientes con FVIII <1 UI/dl o inhibidor >20 UB/ml al
diagnóstico Monitoreo de eventos adversos (glucosa elevada, infección, trastornos psiquiátricos) |
| Corticosteroides y ciclofosfamida | Corticosteroide igual que arriba; ciclofosfamida 1-2 mg/kg VO diario (alternativa ~5 mg/kg IV cada 3-4 sem)* | Puede tener una respuesta más rápida que la
monoterapia con esteroides, pero un mayor perfil de eventos
adversos Asociado con mayor tasa de remisión completa Monitoreo de mielosupresión (plaquetas, glóbulos blancos) e infección |
| Corticosteroides y rituximab | Corticosteroide igual que arriba; rituximab 375 mg/m2 IV semanal x 4 (alternativa 100 mg semanal x 4)* | Rituximab no recomendado como monoterapia inicial a menos que otra TIS esté contraindicada |
*Poca información en HAA, pero reportes disponibles en otras enfermedades autoinmunes.
HAA: hemofilia A adquirida; VO: vía oral; IV: intravenoso; Ig: inmunoglobulina; ; UB: unidades Bethesda; TIS: terapia de inmunosupresión.Adaptada de Kruse-Jarres, et al., 201785.
Consideraciones relevantes de la terapia de inmunosupresión
El esquema de TIS más utilizado es prednisona como monoterapia (1-2 mg/kg/día por 4 a 6 semanas) o en combinación con ciclofosfamida (CFA) por un máximo de 5 semanas, ya sea oral o parenteral, con respuesta entre el 70 y el 80%, sin diferencia significativa en la mediana de tiempo para alcanzar respuesta. De acuerdo con el registro de la cohorte de pacientes EACH2, el uso combinado de esteroide/CFA obtuvo una mayor tasa de respuesta, del 70 vs. 48%, comparado con prednisona como monoterapia, sin diferencia significativa de supervivencia global, justificado por una mayor toxicidad por la CFA. Se ha sugerido la terapia combinada de prednisona/CFA en pacientes con títulos de inhibidor > 100 UB/ml y nivel de FVIII < 1%, o en pacientes con padecimientos autoinmunes.92 El estudio prospectivo de Tiede, et al.93 documentó como valores predictivos independientes de buen pronóstico a pacientes con niveles de FVIII > 1% y títulos de inhibidor < 20 UB/ml, sugiriendo el empleo de una terapia menos agresiva y con menos efectos secundarios en este subgrupo de pacientes.
El uso de anticuerpos monoclonales anti-CD20 (rituximab) se reserva para aquellos casos con una contraindicación absoluta para los fármacos de primera línea y/o como una alternativa de segunda línea o recaída. La tasa de respuesta de los esquemas con rituximab se sitúa entre las cifras obtenidas con esteroide y/o esteroide/CFA en un 59%; sin embargo, el tiempo de respuesta con rituximab como monoterapia es más prolongado que con otros esquemas (mediana de 64 días), pero con una menor tasa de recaídas, del 4 vs. 14 y 19% con esteroide/CFA o esteroide solo, respectivamente. La revisión de Cochrane del 2016 no identificó ningún ensayo clínico controlado que evaluara de manera concluyente el uso de rituximab en esta patología, por lo que sugiere que la toma de decisiones se establezca de acuerdo con estudios observacionales de mayor tamaño y con el menor sesgo metodológico.94
El uso de la inmunoglobulina intravenosa (IgIV) sola o en combinación con esteroides no ha mostrado eficacia como primera línea en diversos estudios, sin embargo, a diferencia de la hemofilia congénita, donde la ITI con dosis altas de FVIII tiene una indicación precisa, el estudio de Zeitler analizó los resultados en 60 de 67 pacientes con HAA (mayoría idiopática) mediante el protocolo modificado de Bonn-Malmö, que combinó la inmunoabsorción con volúmenes plasmáticos altos, IgIV e inmunosupresión con CFA/prednisona y administración posterior de FVIII a dosis de 100 U/kg cada 4 a 6 horas, obteniendo una remisión completa (FVIII ≥ 70% e inhibidor indetectable) del 91% con una mediana de seguimiento de 62 meses.84 A la fecha de la publicación del presente documento, es el único estudio en el que se intenta la inmunotolerancia como fin terapéutico en HAA. Por último, además de la baja incidencia de HAA en población pediátrica, la TIS es controvertida.
Los pacientes que no presenten un incremento en los niveles FVIII posterior a 3-5 semanas de tratamiento deberán ser considerados para recibir una terapia de segunda línea. Las alternativas de TIS de segunda línea incluyen inhibidores de calcineurina como la ciclosporina (útil en HAA asociada a lupus a dosis de 10-15 mg/kg) o el tacrolimús, micofenolato de mofetilo, múltiples fármacos inmunosupresores y citotóxicos (como azatioprina, cladribina, vincristina)85,87 o protocolos de inmunotolerancia, pero con escaso aporte por la literatura médica.
El estudio retrospectivo de Napolitano analizó 105 casos de HAA asociados a alguna neoplasia, donde los tres tumores sólidos más frecuentes fueron de próstata, pulmón y colon (25.3, 15.8 y 9.5% respectivamente), para neoplasias hematológicas: linfoma 24.4%, leucemia linfocítica crónica 22.3% y discrasias de células plasmáticas 20%. Las mejores respuestas (completa o parcial) con erradicación de inhibidor se obtuvieron en pacientes con tratamiento efectivo contra el cáncer, ya sea con quimioterapia o tratamiento quirúrgico (del 88.8%), sin reportar alguna diferencia estadística entre las variables de sexo, edad, títulos de inhibidor, tipo de cáncer y terapia hemostática empleada. Esta revisión señala la interacción entre células neoplásicas y su capacidad de generar una respuesta autoinmune por diversos mecanismos.95
Pronóstico
El pronóstico del paciente con HAA depende de la gravedad y sitio de las manifestaciones hemorrágicas, del acceso al tratamiento específico (hemostático y TIS) y de la presencia de comorbilidades. La tasa de mortalidad general varía entre el 9 y el 33%, dependiendo de las series y fecha de publicación. Las defunciones tempranas por hemorragia ocurren en las primeras semanas posteriores al diagnóstico, con cifras entre el 3.2 y el 9.1%, mientras que las muertes tardías se asocian a efectos secundarios de la TIS. En las series publicadas después del 2010 la mortalidad asociada a sepsis o infección fueron del 12 al 16%.90,93,96 Se ha descrito una mortalidad ≥ 20% en mayores de 65 años y con alguna neoplasia maligna subyacente.97 No existe evidencia contundente referente a los factores pronósticos de respuesta al tratamiento en pacientes con HAA, algunos estudios señalan que pacientes con nivel de FVIII < 1%, título de inhibidor ≥ 20 UB/ml, pobre desempeño físico al diagnóstico y autoanticuerpos de tipo IgA anti-FVIII tienen un peor pronóstico.
La tasa de recaída del inhibidor es del 12 al 18%, principalmente en los dos primeros años, lo cual depende del tipo de TIS utilizada, con una menor tasa para las terapias con rituximab, como se expuso previamente. La recomendación internacional para el seguimiento del paciente que ha obtenido remisión completa (inhibidor indetectable < 0.6 UB/ml y FVIII > 50%) es mediante el monitoreo del TTPa y FVIII mensualmente durante los primeros 6 meses, seguido de cada 2 o 3 meses en el siguiente semestre y posteriormente cada 6 meses. En caso de recaída se ha descrito que el 50% de los casos responde a la misma terapia inicial y una cuarta parte dependerá de esteroides. Un 30% de los pacientes presentan remisión espontánea (desaparición del autoanticuerpo), generalmente en casos de HAA asociada a embarazo o fármacos.
Hemofilia A adquirida y embarazo
La incidencia de HAA es de 1 en 350,000 nacimientos.86 Se presenta habitualmente en el primer embarazo y en los primeros 3 meses posparto, con una presentación clínica similar a otros pacientes con HAA, pero con mayor riesgo de sangrado uterino y/o vaginal. Existe un riesgo latente de hemorragia en el producto por el potencial paso de autoanticuerpos vía transplacentaria.
El inhibidor adquirido en el periodo posparto desaparece de manera espontánea en un 60-100% de los casos, con una media de 30 meses; sin embargo, se debe de iniciar tratamiento inmediato al diagnóstico por el riesgo de sangrado. El tratamiento con agentes puente es similar al de otros pacientes con HAA, la TIS se restringe al uso de esteroide a las dosis establecidas, con una contraindicación del empleo de citotóxicos como la CFA. Un solo centro ha reportado el uso eficaz de rituximab posparto. El pronóstico de la HAA asociada a embarazo es bueno si se detecta y maneja de manera oportuna, además de tener una baja tasa de recaídas en un segundo embarazo.
Hemofilia B adquirida
Se han reportado dos casos de HB adquirida, en el sentido de adquisición de un fenotipo de HB con niveles de factor FIX entre un 5 y un 12% posterior al trasplante hepático ortotópico de donadores con HB no diagnosticados previamente y no por la presencia de autoanticuerpos, como ocurre en la HAA. A diferencia del FVIII, que se produce en diversos sitios extrahepáticos (bazo, riñón y endotelio), el FIX se sintetiza exclusivamente en el hígado, por lo que la transmisión inadvertida de una HB leve vía trasplante hepático debe considerarse en paciente con sangrado y/o una coagulopatía con prolongación del TTPa posterior al trasplante98,99.

