











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome X frágil (SXF) es una condición de repetición de nucleótidos no mendeliana. El SXF se debe a la pérdida de función del gen FMR1 (fragile x mental retardation 1). El gen FMR1 se encuentra en el cromosoma Xq27.3 y codifica la proteína FMRP, cuya función es controlar la traducción de mensajeros específicos. La repetición de tripletes CGG (> 200 repeticiones) y la metilación del promotor conllevan el silenciamiento del gen. Sin embargo, el mecanismo biológico responsable de la presentación del SXF no se conoce del todo. Aproximadamente, 30 % de las niñas y 90 % de los niños afectados con la mutación completa presentan discapacidad intelectual y 60 % de los niños son diagnosticados con trastornos del espectro autista (TEA). Los trastornos de ansiedad ocurren entre 70 y 80 % de las personas con SXF.
La prevalencia más aceptada del SXF es de aproximadamente uno en 5000 hombres y una en 4000 a 8000 mujeres. Sin embargo, aún no hay un consenso global debido a la complejidad del diagnóstico molecular y la variedad en la presentación clínica de quienes no están severamente afectados.1 Se han reportado prevalencias mucho más altas en España2 y Colombia, donde recientemente se reportó un conglomerado genético con la prevalencia más alta del síndrome hasta la fecha;3 así como una casi inexistente en China, donde se especula que la falta de investigación y especialización clínica en áreas del neurodesarrollo es la causa principal de la escasez diagnóstica del síndrome.
Bases biológicas del síndrome X frágil
El mecanismo biológico exacto responsable de la presentación del SXF no se conoce, sin embargo, se sabe que reside en la capacidad de la proteína FMRP de unión a ARN y a proteínas. Específicamente, FMRP se une a ribosomas y está presente en los compartimentos sinápticos, donde controla la traducción de mensajeros específicos. La pérdida de FMRP produce alteraciones de la conectividad sináptica en las neuronas, que se traducen en los síntomas específicos del SXF. Estas alteraciones de la conectividad sináptica se ponen de manifiesto en el cerebro con la disminución de la cantidad de dendritas y espinas en las neuronas de pacientes con SXF.
La falta de FMRP en neuronas conlleva una expresión exacerbada de receptores de glutamato, tanto metabotrópicos (mGluR5) como ionotrópicos (AMPA y NMDA).4 Las proteínas de síntesis, degradación y trasporte del ácido g-aminobutírico (GABA) y los receptores de GABA también están reducidas.5 No se sabe exactamente cómo estos cambios en los sistemas de neurotransmisores afectan la morfología de dendritas y espinas en las neuronas, pero se sospecha que están relacionadas. El papel de la proteína FMRP en las células gliales es menos conocido, pero se sabe que en SXF la proteína FMRP regula la traducción de mGluR5 en astrocitos6 y la producción de mielina en oligodendrocitos.7 Durante el desarrollo prenatal, las células de la glía radial contienen FMRP, que interviene en el transporte activo de ARN mensajero a lo largo de la fibra glial.8 Un cambio en cualquiera de estos mecanismos puede colaborar al desarrollo de las alteraciones cognitivas en los pacientes con SXF.
FMRP ha sido relacionada con la regulación de canales iónicos. FMRP se une al C-terminal de los canales Slack, activados por potasio. La activación de los canales Slack contribuye a los patrones de activación de una gran variedad de neuronas, lo que sugiere que las alteraciones observadas en el SXF pueden ser generadas por patrones de actividad alterados.9 A su vez, FMRP también puede regular la liberación de neurotransmisores a través de la modulación del potencial de acción vía canales de potasio de larga conducción activados por calcio (canales BK).10
La presencia de una pequeña fracción de FMRP en el núcleo celular indica que dicha proteína puede tener funciones previamente no reconocidas. De hecho, varios estudios han desvelado funciones relacionadas con la expresión del ADN y la función genómica, como en la estabilización del ADN, la regulación epigenética del ADN, la regulación del ARN nuclear y la respuesta al daño del ADN.11
La proteína precursora del b amiloide (APP) también ha sido relacionada con el SXF, a través de un mecanismo dependiente del receptor mGluR5. APP es procesada por secretasas que producen b amiloide (Ab), péptido predominante en las placas seniles en la enfermedad de Alzheimer.12
Presentación clínica
Los afectados con la mutación completa del gen FMR1 presentan características fenotípicas especiales que incluyen cara alargada, orejas grandes y prominentes, hipermovilidad articular y macroorquidismo.13 Más de 90 % de los niños afectados presentan retraso del desarrollo y aproximadamente el 50-60 % son diagnosticados con TEA.14 Durante el transcurso de su vida, tanto hombres como mujeres presentan alteraciones del comportamiento comúnmente asociadas con el síndrome, usualmente de inicio durante la infancia: la ansiedad y el trastorno de atención e hiperactividad (TDAH) son las más prevalentes; aunque los desórdenes compulsivos como la hiperfagia y la agresividad también son comunes (Tabla 1). Además de las alteraciones comportamentales y los problemas de aprendizaje y adaptación social, de 15 a 20 % de los pacientes con SXF presentan convulsiones, más prevalentes en aquellos con autismo; más de 30 % tiene problemas de obesidad, alteraciones del sueño y alguna disfunción gastrointestinal, incluyendo reflujo gastroesofágico. El estrabismo y la otitis media recurrente son problemas comunes durante la primera infancia.
Tabla 1 Características clínicas
| Características clínicas | Prevalencia | |
|---|---|---|
| Físicas | Cara alargada | 83 % más común en adultos |
| Macrocefalia | 50-81 % | |
| Orejas prominentes | 75 % | |
| Mandíbula prominente | 80 % en adultos | |
| Pies planos | 29-69 % | |
| Macroorquidismo | 95 % desde la adolescencia | |
| Hipermovilidad articular | 50-70 % más común en niños | |
| Psicológicas/psiquiátricas | TDAH | 80 % niños y 40 % niñas |
| TEA | 50-60 % niños y 20 % niñas | |
| Ansiedad | 58-86 % | |
| Agresividad | 40 % niños y 10-15 % niñas | |
| Desarrollo | Discapacidad intelectual | 85 % niños y 25-30 % niñas |
| Déficit del lenguaje | 100 % niños y 60-75 % niñas | |
| Otros | Estrabismo | 8-30 % |
| Otitis recurrente | 50-75 % en la infancia | |
| Problemas gastrointestinales | 30 % | |
| Obesidad | 30-60 % | |
| Convulsiones | 15-20 % |
Adaptado de referencia 14. TDAH = trastorno de atención e hiperactividad, TEA = trastornos del espectro autista.
El fenotipo tiene algunas variantes. Los más afectados con la mutación son los hombres; las mujeres presentan un fenotipo atenuado por el índice de activación del segundo cromosoma X no afectado. Más de 70 % de las mujeres tiene un coeficiente intelectual bajo, si bien considerado promedio comparado con la población general y en menor proporción comparado con los hombres que presentan problemas de lenguaje.13 La segunda variante son los mosaicos, que presentan algunas líneas celulares con la mutación completa y otras en el rango de premutación, lo que los expone al riesgo de padecer los problemas propios de la premutación como el síndrome de temblor/ataxia (FXTAS);15,16 o algunas líneas celulares con metilación y, por ende, el gen silenciado y otras sin metilación, en este caso los afectados también presentan menor grado de afectación cognitiva.17
Además de las características fenotípicas comúnmente reconocidas, los afectados presentan anomalías del tejido conectivo, atribuidas a que FMRP regula componentes esenciales de la matriz extracelular. Además de las alteraciones musculoesqueléticas más comunes, como la hiperextensión de las articulaciones metacarpofalángicas, los pies planos y escoliosis, se han descrito alteraciones en los sistemas cardiovascular y genitourinario.18
Imágenes de resonancia magnética del cerebro de los pacientes con SXF demuestran que por lo general el cerebro es más grande de lo normal y con incremento en el tamaño de los ventrículos laterales. El vermis cerebelar presenta hipoplasia, una de las características más representativa, que puede ir acompañada de la reducción del cerebelo entero y alteraciones de los pedúnculos cerebelosos. Además, el núcleo caudado, sobre todo la cabeza, es más grande, principalmente en hombres. El hipocampo también está agrandado en pacientes jóvenes. Por el contrario, la ínsula y la amígdala son más pequeñas. Asimismo, el fascículo uncinado también presenta alteraciones de la materia blanca.13
Interacción entre SXF, autismo y trastorno de déficit de atención e hiperactividad
Existe una relación estrecha entre la presencia de SXF, TEA y TDAH. Aproximadamente 2 % de todos los casos diagnosticados de trastornos del espectro autista (TEA) son atribuibles al SXF; mientras que más de 60 % de los niños con SXF son diagnosticados con TDAH, TEA o los dos. El SXF es la principal causa genética conocida de los TEA, sin embargo, solo 20 % de los casos de autismo son reconocidos como el resultado de mutaciones monogénicas y solo 2 a 6 % se debe a la mutación del gen FMR1. Los afectados por las dos morbilidades, como sucede en 50 a 60 % de los niños y 20 % de las niñas con SXF, presentan un compromiso más severo tanto del déficit cognitivo y del lenguaje como en los problemas del comportamiento.19 Los ensayos clínicos controlados han demostrado que a pesar de que SXF y TEA comparten sintomatología psiquiátrica, los afectados no responden con la misma eficacia a tratamientos específicos,20,21 por lo cual se sugiere que los mismos síntomas emergen de distintos mecanismos.
Diagnóstico
La edad aproximada del diagnóstico del SXF es de 36 meses,22 a pesar de que la mayoría de los padres reporta reconocer algún tipo de retraso en el neurodesarrollo durante el primer año de vida. El tamizaje de poblaciones de alto riesgo puede realizarse por prueba de reacción en cadena de la polimerasa (PCR), de relativo bajo costo que requiere una sola gota sanguínea. El método utiliza un cebador quimérico que apunta aleatoriamente dentro de la región ampliada de CGG en el gen FMR1.23 Este método se ha utilizado exitosamente en varios estudios poblacionales.2,3 La prueba confirmatoria del diagnóstico es Southern Blot. Tejada hizo la evaluación completa de las ventajas y controversias de la prevención del SXF haciendo uso del diagnóstico prenatal.24 En 2017, Riley y Wheeler describieron la problemática del establecimiento del tamizaje posnatal en Estados Unidos.25 La recomendación actual de la Academia Americana de Pediatría es realizar pruebas genéticas a los niños con discapacidad intelectual o retraso global del desarrollo.26 De encontrarse un caso nuevo de SXF debe realizarse diagnóstico en cascada a todos los miembros de la familia inmediata, con el fin de identificar a los portadores de la premutación que tienen el potencial de expandir la mutación completa a sus descendientes (Figura 1) . Recientemente Lubala et al. realizaron un metaanálisis en el que se incluyeron 10 estudios de tamizaje y se propuso una puntuación clínica para las siete características más específicas del SXF; esta lista toma en consideración las diferencias, sobre todo faciales, que pueden encontrarse en diferentes grupos étnicos. Esta herramienta clínica es de suma importancia en áreas donde no todos los afectados con discapacidad intelectual o TEA pueden someterse a pruebas genéticas debido a limitación de los recursos (Tabla 2).27
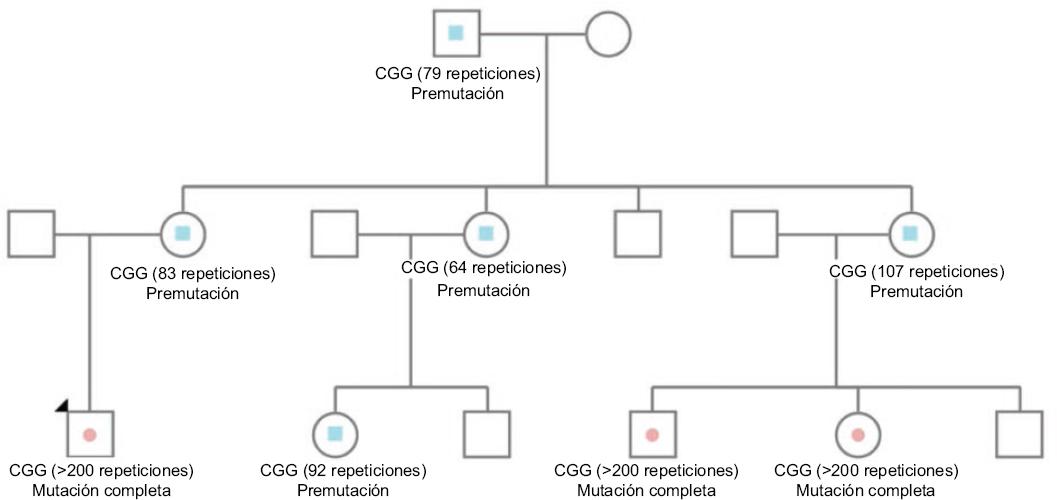
Figura 1 Árbol familiar. Al hacer el diagnóstico molecular se recomienda realizar diagnóstico en cascada a todos los miembros de la familia inmediata. El hombre portador de la premutación en la primera generación pasa la premutación a 100 % de sus hijas mujeres, mientras que sus hijos varones no serán portadores de la premutación o de la mutación completa. Los hijos e hijas de las mujeres en la segunda generación tienen 50 % de probabilidad de tener la premutación o la mutación completa. Solo las mujeres con la premutación tienen la capacidad de expandir la mutación completa tanto a sus hijos varones como mujeres y estos de presentar el SXF en la tercera generación.
Tabla 2 Lista de verificación clínica para el síndrome X frágil
| Característica | Puntuación | |
|---|---|---|
| 1 | 2 | |
| Piel suave y aterciopelada en las palmas de las manos con exceso de piel en el dorso de la mano | X | |
| Pies planos | X | |
| Orejas grandes y prominentes | X | |
| Pliegue plantar | X | |
| Macroorquidismo* | X | |
| Historia familiar de discapacidad intelectual | X | |
| Comportamiento autista | X | |
| Total | 4 | 6 |
*Varones después de la pubertad. La puntuación máxima es de 10 puntos para varones después de la pubertad y de nueve para varones antes de la pubertad o mujeres. En pacientes con puntuación superior a 5 debe considerarse el diagnóstico molecular de SXF. Adaptado de referencia 27.
Diagnóstico en países hispanos
A pesar de la recomendación de realizar pruebas genéticas en niños con discapacidad intelectual o retraso global del desarrollo y en aquellos cuyas familias están afectadas, estas pruebas no se realizan en numerosos países hispanos. Las pruebas genéticas diagnósticas están disponibles y varios países en Latinoamérica han reportado estudios de prevalencia del SXF, sin embargo, determinar la prevalencia real de los desórdenes genéticos es difícil debido a que en numerosas naciones hispanas no existe un registro oficial nacional. Países como Chile, Brasil, Colombia, Argentina, Perú y España han hecho visible la necesidad de implementar mejores procesos de tamizaje y diagnóstico de enfermedades genéticas prevalentes, incluyendo del SXF. Además, hay barreras económicas, políticas y sociales que enfrenta el campo neurogenético, principalmente en los países en desarrollo.2,3 Hasta la actualidad, el diagnóstico del SXF se basa principalmente en los hallazgos fenotípicos, con la posibilidad de realizar pruebas genéticas por recomendación del especialista. En muchos casos no se realizan debido a su alto costo, a que no están cubiertas por los seguros de salud y, en otros casos, a la poca disponibilidad de laboratorios certificados para realizar el análisis de ADN en muestras sanguíneas.
Tratamiento
No hay cura para el SXF, por lo que el tratamiento se limita al control de los síntomas asociados. Actualmente las líneas de investigación se centran en desarrollar tratamientos eficaces para los distintos problemas psiquiátricos y cognitivos que padecen los afectados (Tabla 3). En 2017, Gantois et al. investigaron la eficacia de metformina como modulador de la cascada de mGluR/mTORC1-ERK en modelos animales de SXF; reportaron mejoría en el comportamiento social y cognitivo, así como en las anormalidades morfológicas (disgenesia de la espinas dendríticas en el hipocampo) y electrofisiológicas (depresión a largo plazo).28 Estos hallazgos motivaron el inicio de la investigación del tratamiento con metformina en la práctica clínica. El primer reporte demostró beneficio principalmente en comportamientos problemáticos como irritabilidad, agresividad y evasión social en pacientes adultos con SXF, además de beneficiar el control del apetito y el peso en quienes presentaban el fenotipo de Prader-Willi.29 Por esta razón, en la actualidad estudios controlados tanto en Estados Unidos como en Canadá buscan determinar la eficacia de metformina como tratamiento de este síndrome.
Tabla 3 Medicamentos con eficacia en el tratamiento de síndrome X frágil
| Medicamento | Dosis máxima/día | Efectos adversos comunes |
|---|---|---|
| Metformina | 1000 mg < 50 kg 2000 mg > 50 kg | Náusea, diarrea, cefalea, pérdida de peso |
| Sertralina | 2.5 a 5.0 mg niños de 2 a 6 años 10 a 100 mg niños mayores de 6 años y adolescentes | Diarrea, pérdida de apetito, hiperhidrosis, temblor |
| Minociclina | 25 mg < 25 kg 50 mg 25-45 kg 100 mg > 45 kg | Náusea, diarrea, cefalea, mareo, pérdida de apetito, decoloración dental y de la cavidad oral, erupciones cutáneas |
| Lovastatina | 40 mg | Debilidad, síntomas gastrointestinales, molestias musculares, mareo, cefalea, irritabilidad |
| Acamprosato | 1332 mg < 50 kg 1998 mg > 50 kg | Irritabilidad, síntomas depresivos, aumento del comportamiento repetitivo, síntomas gastrointestinales, incluyendo diarrea y estreñimiento |
Adaptado de referencias 21,29,33 y 38.
La sertralina es un medicamento de primera línea para el manejo de la depresión y la ansiedad. Fue estudiado por su potencial beneficio en el lenguaje, sin embargo, demostró mejores resultados en habilidades perceptuales motoras y visuales y participación social en SXF.21
La minociclina también se considera un tratamiento beneficioso en SXF. Se conoce que reduce los niveles de la matriz metalopeptidasa 9 (MMP-9),30 endopeptidasa dependiente de cinc encargada de regular la actividad sináptica crítica para el desarrollo y la plasticidad del sistema nervioso central.31 Su inhibición se produce por la unión con FMRP, proteína que se encuentra ausente en el SXF. Los problemas de regulación de MMP-9 se consideran parte de la fisiopatología no solo de los problemas de aprendizaje, sino también de las anomalías encontradas en el tejido conectivo.18
El acamprosato, un antagonista del receptor mGluR5, modificó el comportamiento ansioso y las pruebas locomotoras en el modelo animal de SXF32 y demostró mejoría en áreas de comportamiento social e hiperactividad en pacientes pediátricos con TEA y SXF.33 Debe considerarse un medicamento beneficioso para el manejo de los pacientes con SXF y problemas de adicción al alcohol.34
Los estudios de tratamiento con lovastatina en modelos animales de SXF postulan este medicamento como un tratamiento profiláctico para la epileptogénesis y sugieren que podría mejorar las funciones sensoriales y cognitivas.35 Los ensayos clínicos no controlados demostraron buena tolerancia en el tratamiento con pocos efectos adversos y reportaron beneficios tanto en el comportamiento como en las habilidades adaptativas.36 A nivel molecular se demostró que los cambios en la fosforilación de la cinasa regulada por señal extracelular (ERK) se relacionan con la respuesta clínica a lovastatina.37
Existen otros medicamentos que pueden mejorar los sistemas neurobiológicos en SXF que no se consideran tratamientos específicos para el síndrome, sino que ayudan a controlar las características psiquiátricas más comunes. Estos incluyen los estimulantes (metilfenidato y anfetaminas) y atomoxetina, que pueden mejorar los síntomas del trastorno de atención e hiperactividad, por lo general, en niños mayores de cinco años; también se pueden usar los agonistas alfa adrenérgicos (guanfacina o clonidina) antes de los cinco años para calmar la hiperactividad. La clonidina es especialmente eficaz para mejorar los trastornos del sueño, de no tener una buena respuesta al tratamiento con melatonina. Para el manejo de la agresividad o los desórdenes del estado de ánimo, los antipsicóticos (risperidona o aripiprazol) son adecuados, pero pueden causar aumento de peso.
Conclusión
Los individuos afectados con el SXF presentan discapacidad intelectual, TEA y TDAH. Aunque existen muchos medicamentos para el manejo de las comorbilidades comunes, no hay tratamientos específicos. El objetivo del tratamiento temprano es mejorar la discapacidad intelectual, las dificultades de la comunicación y la interacción social característicos del SXF. Además, a pesar de la recomendación de realizar pruebas genéticas en niños con discapacidad intelectual o retraso global del desarrollo, estas no se realizan en muchos de los países latinoamericanos. Es de mayor importancia implementar el análisis SXF en todos los países hispanos.

