











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Common variable immunodeficiency (CVID) is the most common primary symptomatic immunodeficiency in the world.1 Its diagnosis is based on the criteria proposed by the American and European immunodeficiency societies: a decrease in immunoglobulin G (IgG) serum levels below two standard deviations for age and decrease in other serum immunoglobulin (IgA or IgM), age older than two years, absence of isohemagglutinins or poor response to vaccines and exclusion of other causes of hypogammaglobulinemia.1
The main clinical manifestation in CVID is infectious; however, other complications such as autoimmunity, gastrointestinal disorders, granulomatosis, lymphoproliferation and neoplasms are common.1-3 The characteristic immune alteration is hypogammaglobulinemia with B lymphocyte involvement; however, involvement includes other cells such as CD4+ T lymphocytes, regulatory lymphocytes (Treg CD25+ FoxP3+ lymphocytes), CD8 T lymphocytes, monocytes, antigen-presenting cells and natural killer (NK) cells.1,2,4 CVID has been associated with various genetic and environmental factors; however, none of them has been able to fully explain the multiple clinical manifestations, severity and evolution of the disease.1,2
Human intestinal microbiota has been proposed as a key micro-ecosystem in the health-disease process, where dysbiosis is associated with obesity, infections, autoimmunity and allergic diseases.5-7 Gut microbiota evolution, development and diversity starts since birth and is modified throughout life.8 Ninety percent of the microorganisms that colonize the human intestine are represented by Firmicutes, Bacteroides, Actinobacteria, Fusobacteria, Protebacteria and Verrucomicrobia phyla.9,10 This wide bacterial diversity has important functions in the development of the innate and adaptive immune system in a bi-directional feedback process.7,11,12 Notwithstanding, the study of intestinal microbiota in primary immunodeficiencies is limited. Considering the amount of activities of microbiota in immune regulation and the various immune alterations of CVID, there is a need to delve into the knowledge of intestinal microbiology in patients with this medical condition. Therefore, the first analysis of the gut microbiota in Mexican patients with CVID is presented, where the structure of the intestinal microbial community associated with this pathology is described.
Method
During 2016, fecal samples were collected from five patients older than 18 years who had been diagnosed with CVID and who were under the care of the Department of Clinical Immunology and Allergy of the National Medical Center of the West, Mexican Institute of Social Security, in Guadalajara, Jalisco, Mexico. The protocol was approved by the ethics committee of that institution.
A stool sample per patient was collected using a sterile container; 0.25 g of each sample was placed in BashingBead™ lysis tubes with 750 µL of Xpedition™ Zymo Research™ lysate buffer/stabilizer. Each tube was placed in a cell disruptor (TerraLyzer™) for 30 seconds for DNA preservation.
DNA was extracted from the samples using the Soil/Fecal DNA MiniPrep commercial kit (Zymo Research™). The extracted DNA was run on 1.2 % agarose gel at 80 V for 45 minutes in a BIO-RAD electrophoresis chamber to visualize the presence of high molecular weight DNA. Visualization was carried out in a GelMax™photodocumenter (UVP®). The amount of DNA per sample was measured on a Qubit® fluorometer. Amplification of the V3 and V4 regions of the 16S rRNA gene was carried out, using the S-D-Bact-0341-b-S-17, 5´-CCTACGGGNGGCWGCAG-3´ and SD-Bact-0785-aA-21, 5´-GACTACHVGGGTATCTAATCC-3´ primers, which produce an amplicon of approximately 460 bp.13 These sequences were synthesized with the overhang adapters of the Illumina protocol (2017a), which produce an amplicon of approximately 550 bp.
The Illumina PCR protocol was used with 12.5 µL of MyTaq™ Ready Mix 1X (Bioline®), 1 µL of each primer (10 µM), 5 µL of DNA (50 ng total) and 5.5 µL of molecular grade H2O. The following cycle was used: 95 °C for three minutes; 25 cycles at 95 °C for 30 seconds, 55 °C for 30 seconds, 72 °C for 30 seconds; 72 °C for five minutes in a Labnet Multigene™ Gradient PCR thermal cycler. 1 µL of the PCR products was placed on a Bioanalyzer DNA 1000 chip to verify the size of the amplicon (approximately 550 bp). Amplicon purification was carried out with 0.8 % Agentcourt® AMPure® XP beads. Subsequently, the amplicons were labeled with Nextera XT Index Kit™ for the creation of the libraries, using 25 µL of MyTaq™ Ready Mix 1X (Bioline®), 5 µL of each primer (N7xx and S5xx), 5 µL of DNA and 10 µL of molecular grade H2O. The following cycle was used: 95 °C for three minutes; 10 cycles at 95 °C for 30 seconds, 55 °C for 30 seconds, 72 °C for 30 seconds, 72 °C for five minutes. Purification of the libraries was carried out with 1.2 % Agencourt® AMPure® XP beads. 1 µL of the final library of some randomly selected PCR products was placed on a DNA 1000 Bioanalyzer chip to verify the size of the amplicon, with a size of approximately 630 bp being expected. Finally, quantification, normalization (equimolarity), library grouping and next-generation mass sequencing (MiSeq Illumina® of 2x250 paired-end readings) were performed, according to the Illumina 16S protocol for metagenomics.
Sequence analysis was performed in Oracle VM VirtualBox 5.1.14 on the MGLinux platform using the Quantitative Insights Into Microbial Ecology bioinformatics software, version 1.9.0.14 The process was initiated by assembling the forward and reverse sequences of the samples, using the PEAR15 program with an overlap of 50 bp, a minimum length of 430 bp per reading and maximum length of 470 bp, a quality criterion Q30 (one erroneous base per 1000 bases) and a p-value < 0.0001. The files were next converted into the FASTA format and elimination of chimeric sequences from the samples was carried out with USEARCH.16 Operational taxonomic units (OTU) selection was carried out using the UCLUST16 method at 97 % of similarity; a representative sequence was obtained for each OTU and taxonomy was assigned taking the EzBioCloud databases as reference.17 The OTU table was constructed in the Biological Observation Matrix (BIOM) format,18 the domains were separated and singletons were filtered (OTUs that only had one observation).19 OTU absolute abundance tables were obtained and the number of sequences was plotted by the number of taxa at the genus level, to confirm an adequate coverage depth (asymptotic trend curves); this graph was generated using PAST, version 3.15.20 The number of sequences that were simultaneously reached by all samples was 80,000, and a simple random rarefaction process was carried out,21 taking this value as the number of sequences to be generated. This way, a standardized BIOM file was obtained for all samples. The phylum, class, order, family and genus taxonomic levels relative abundance was obtained; the phyla and genera were plotted in Excel.
Results
Total sequences mean obtained for all patients prior to assembly was 217,381, with a mean of assembled sequences of 170,777; mean quality sequences after chimera removal was 162,137. After taxonomic assignment, a mean of 157,355 bacterial sequences was obtained, which after singletons were eliminated was 110,265. Mean OTU was 5679 (Table 1). Depth of coverage in terms of the number of bacterial taxa per patient was adequate, since asymptotic tendency was achieved in each curve (Fig. 1).
Table 1 Information on the bacterial sequences used in the present study
| Patient | TS | AS | DS | QS | BS | BSSE | OTU |
|---|---|---|---|---|---|---|---|
| P1 | 199 924 | 188 675 | 11 249 | 184 796 | 178 145 | 120 665 | 6 442 |
| P2 | 228 803 | 189 333 | 39 470 | 165 517 | 106 264 | 106 264 | 5 401 |
| P3 | 190 836 | 143 260 | 47 576 | 132 649 | 127 663 | 84 229 | 5.087 |
| P4 | 233 012 | 177 792 | 55 220 | 175 753 | 172 519 | 131 572 | 4 831 |
| P5 | 234 330 | 154 823 | 79 507 | 151 499 | 147 932 | 108 594 | 6 636 |
| Mean | 217 381 | 170 777 | 46 604 | 162 137 | 157 355 | 110 265 | 5 679 |
TS = total number of sequences, AS = number of assembled sequences, DS = number of discarded sequences, QS = quality sequences after chimera elimination, BS = bacterial sequences after taxonomic assignment, BSSE = bacterial sequences after singleton elimination, OTU = operational taxonomic units.
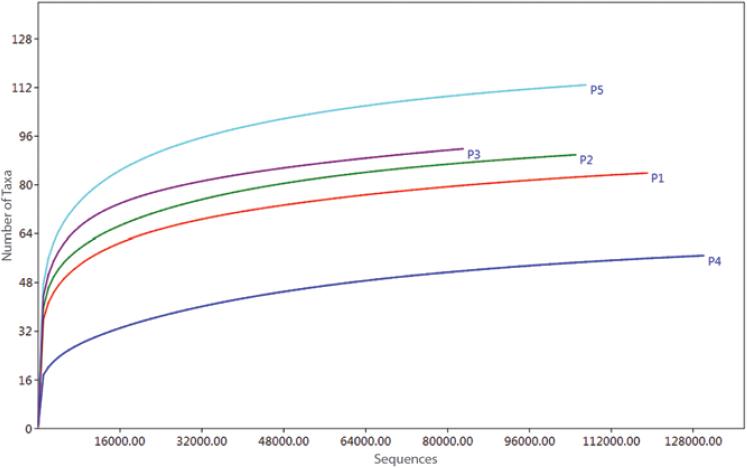
Figure 1 Depth of coverage of fecal bacterial sequences of patients with common variable immunodeficiency.
Ten phyla were recorded, out of which Firmicutes was the most abundant (x¯ = 89 %), with Actinobacteria (x¯ = 6 %) and Verrucomicrobia (x¯ = 3 %) following in dominance (Fig. 2). Fifteen classes were determined, with Clostridia being the most abundant (x¯ = 89 %), followed by an unidentified class of the Actinobacteria phylum (x¯ = 6 %) and by the Verrucomicrobia class (x¯ = 3 %).
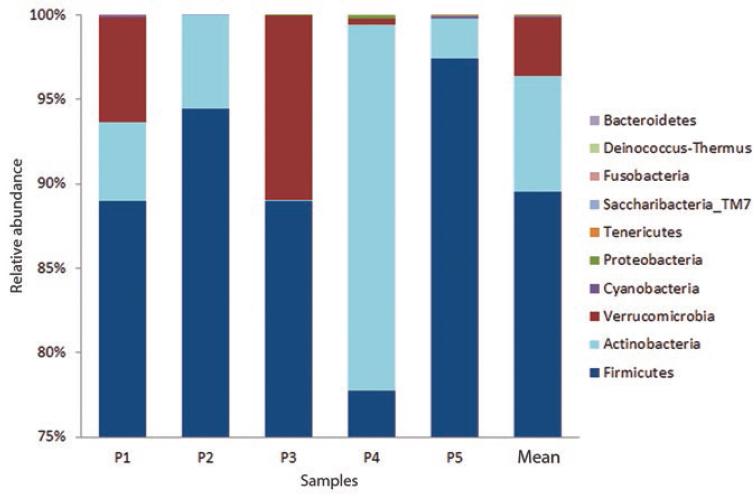
Figure 2 Relative abundance of fecal bacteria phyla of patients with common variable immunodeficiency; the last bar shows average abundance.
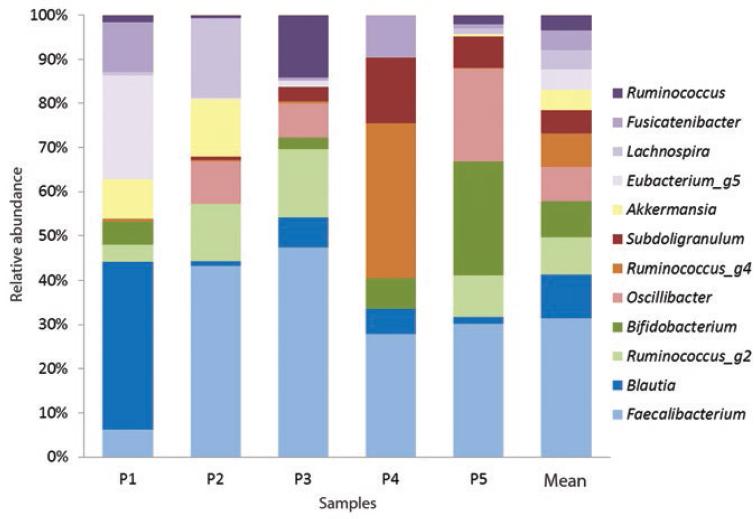
Twenty-one orders were obtained, with Clostridial orders being the most abundant (x¯ = 89 %), followed by Bifidobacterial (x¯ = 6 %) and Verrucomicrobial (x¯ = 6 %). Of the 34 recorded families, Ruminococcaceae (x¯ = 48 %), Lachnospiraceae (x¯ = 39 %), Bifidobacteriaceae (x¯ = 6 %) and Akkermansiaceae (x¯ = 3 %) predominated. One-hundred and sixty-six genera were reported, among which the most abundant were Faecalibacterium (x¯ = 23 %), Blautia (x¯ = 7 %), Ruminococcus g2 (x¯ = 6.3 %) and Bifidobacterium (x¯ = 6 %) (Figure 3). The identified species were Bifidobacterium longum (only species present in 100 % of individuals), Bifidobacterium thermacidophilum (20 % of patients), Propionibacterium acnes (20 %), Eubacterium hallii (20 %) and Fusobacterium nucleatum (40 %).
Discussion
This first approach to the study of gut microbiota in Mexican patients with CVID differs from others reported in healthy subjects, in whom intestinal microbiota has been reported to be mainly composed of Firmicutes and Bacteroidetes, and in less abundance of Actinobacteria, Fusobacteria, Proteobacteria, Verrucomicrobia and Cyanobacteria.5,9,10,22,23
Firmicutes, the most abundant phylum in human gut microbiota,9,23 accounted for almost 90 % of total relative abundance in the present study. Derived from this phylum, the Clostridia class, the Ruminococcaceae and Lachnospiraceae families, eight of the main bacterial genera (Faecalibacterium, Blautia, Ruminococcus, Oscillibacter, Subdoligranulum, Eubacterium, Lachnospira and Fusicatenibacter) and the Eubacterium hallii species were identified.
Clostridium IV and XIVa classes have important immune functions; their main activity is based on the production of short-chain fatty acids from dietary fiber fermentation.24,25 These short-chain fatty acids bind to G-protein-coupled receptors for free fatty acids: GPR43/FFAR2, GPRA1/FFAR3 and GPR109A, which are present in various immune cells;26 their binding stimulates interleukin-10-producing Treg CD25+ FOXP3+ and transforming growth factor beta-naïve T lymphocyte differentiation.24,26 These cytokines favor the differentiation of B lymphocytes towards IgA-producing subtypes.25,27-29
The Actinobacteria phylum was the second in abundance; it includes the Actinobacteria class, the Bifidobacterial order and the Bifidobacterium genus. From this phylum, the Bifidobacterium Longum, Bifidobacterium Thermacidophilum and Propionibacterium Acnes species were identified. Bifidobacterium Longum, which was found in 100 % of individuals with CVID, has probiotic activities that comprise fermenting oligosaccharides, metabolizing complex carbohydrates and participating in the biosynthesis of capsular polysaccharides that interact with Toll-2 type receptors (TLR-2) at the beginning of the innate immune response.29,30 Propinobacterium acnes ferments lactose into propionic acid, which has an antimicrobial effect.30-32 Bifidobacterium thermacidophilum has not been previously reported as part of human intestinal microbiota, however, this result should be taken with caution, since it was recorded only in one patient.
The Verrucomicrobia phylum was the third in abundance; from it, the Akkermansiaceae family and the Akkermansia genus are derived. In most studies conducted in healthy subjects (except for some in Latin American countries33), Verrucomicrobia is not found with high abundance; however, it was one of the most abundant in individuals with CVID. Its main immune function is to maintain the integrity of the intestinal mucus layer and produce antimicrobial peptides.33,34
The low abundance of the Bacteroidetes phylum (x¯ = 0.00001) in the studied patients stands out. These gram-negative bacilli are important in the metabolism of complex polysaccharides that arrive intact to the colon.35
The Firmicutes: Bacteroidetes ratio has been proposed as an indicator of dysbiosis.36-38 It is clear that the difference, and therefore the Firmicutes: Bacteroidetes ratio in patients with CVID of this study are extremely high, a finding that leads to suggest intestinal dysbiosis.
In conclusion, gut microbiota composition is reported for the first time in five Mexican patients with CVID. Although most identified bacteria have a positive interaction with the immune system, the high abundance of Firmicutes and the low presence of Bacteroidetes suggest intestinal dysbiosis in these patients. Although the sample size is small, the reported findings are expected to be the first step to delve into the study of microbiota in its relationship with the immune system. It is considered that the development of an additional multicenter study would be useful to reinforce and deepen the described findings.

