










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta médica de México
versión On-line ISSN 2696-1288versión impresa ISSN 0016-3813
Gac. Méd. Méx vol.142 no.5 Ciudad de México sep./oct. 2006
Artículo de revisión
Fisiopatología de las enfermedades por priones
Physiopathology of prion diseases
Alejandra Mandujano,a Saraí Montes,b Aída Guzman,c Blanca Espinosa,d Daniel Rembao,b Salvador Martínez–Cairo,e Edgar Zentenoc y Jorge Guevarab*
a Facultad de Ciencias. Universidad Autónoma del Estado de México, México
b Laboratorio de Enfermedades Neurodegenerativas y Departamento de Neuropatología. Instituto Nacional de Neurología y Neurocirugía Dr. Manuel Velazco Suárez, México D. F., México
c ENP 1 y Departamento de Bioquímica, Fac. de Medicina, Universidad Nacional Autónoma de México, México D. F., México
d Departamento de Bioquímica, Instituto Nacional de Especialidades Respiratorias, México D. F., México
e División de Investigación, CMNS XXI, IMSS, México D. F., México
*Correspondencia y solicitud de sobretiros:
Dr. Jorge Guevara, Laboratorio de Enfermedades Neurodegenerativas
Instituto Nacional de Neurología y Neurocirugía Manuel Velasco Suárez.
Insurgentes Sur 3877, La Fama, 14269 Tlalpan, México D.F., México.
Correo electrónico: jguevara@innn.edu.mx
Recibido en su versión modificada: 17 de abril de 2006
Aceptado: 12 de mayo de 2006
Resumen
Las enfermedades por priones, son trastornos neurodegenerativos progresivos rápidos e invariablemente fatales que afectan tanto a seres humanos como a animales. Tienenformas de presentación esporádica, genética e infecciosa. Los priones son proteínas celulares. No contienen ácidos nucleicosy no son virus o microorganismos. En todos los casos, provocan muerte neuronal, espongiosis común del cerebro, que caracteriza a estas enfermedades, así como agregación de la proteína amiloide prión en forma de placa. La teoría más importante hasta el momento, es la que trata de explicar el cambio de conformación de la pro teína prión para producir copias de sí misma y para su agregación y lamuerte de lasneuronas. Sin embargo, nuevas formas de explicación toman auge actualmente. Una de las más importantes se basa en entender el contenido y cambio de la glicosilación de la proteína prión patológica. Esto permite explicar algunas de sus interacciones, para entender el cambio de conformacióny las propiedades físico–químicas de la proteína. Así como algunas de las primeras funciones biológicas (como transportador de iones Cu++2) descritas para esta molécula. En esta revisión abordamos todos los tópicos importantes acerca de estas patologías por demás fascinantes.
Palabras clave: Prión, enfermedades por priones, glicosilación, cerebro
Summary
Prion diseases are a group of degenerative disorders characterized by being progressive, fast growing, and fatal, they affect humans and animals. Due to their physiopathogeny, these disorders can be sporadic, genetic, or infectious. Prions are cellular proteins that lack nucleic acids; they are notviruses or microorganisms. Prions induce neuronal death, brain spongiosis, which are a hallmark of these diseases, as well as amyloid prion protein plaque aggregates. Although the causes that favor pathogenic prion proteins remain uncertain, it is possible that conformational changes of the prion protein allow them to create copies of themselves to form aggregates and induce neuronal death. Other theories suggest that quantitative and qualitative changes in the glycosylation pattern induce the pathological prion form. The latter allows to explain some of their interactions and to understand better the conformational changes and the physico–chemical properties of the prion protein. We review some of the first biological functions (as a transporter of Cu2+ ions) that have been described to this molecule. The present review focuses on different aspects of prion diseases aimed at understanding better their physiopathogenic characteristics.
Key words: Prion, prion diseases, glycosylation, brain
Enfermedades causadas por priones
Antes conocidas como Encefalopatías Espongiformes Transmisibles (EETs), las enfermedades causadas por priones o prionopatías (EPRs), son trastornos neurodegenerativos progresivos e invariablemente fatales que afectan tanto a seres humanos como a animales.1,2 Estas presentan formas etiológicas distintas: hereditarias, esporádicas y, a diferencia del resto de las enfermedades neurodegenerativas reportadas hasta ahora, también formas infecciosas. Los tiempos de incubación de las EPRs son muy variables, desde unos cuantos meses hasta años, o incluso, décadas.3,4 En los humanos, las prionopatías presentan un cuadro patológico que incluye ataxia, temblor generalizado, pérdida de coordinación, alteraciones de memoria, disfunción motora, pérdida de las habilidades cognitivas, demencia progresiva e invariablemente, la muerte.3,5 En este tipo de padecimientos se encuentran la enfermedad de Creutzfeldt–Jakob (ECJ), el síndrome de Gerstmann–Straussler–Scheinker (GSS), el Insomnio Familiar Fatal (IFF), la Angiopatía Amiloide Cerebral causada por Priones (AAC–PRP), el kuru y una nueva variante de la enfermedad de Creutzfeldt–Jakob (nvECJ)2,3 (Cuadro I). En los animales, los priones provocan las Encefalopatías Espongiformes del ganado Bovino (EEB) —mejor conocida como "enfermedad de las vacas locas"—, encefalopatías del ganado ovino, caprino, de ungulados como el venado y el alce, así como de felinos silvestres y domésticos. También causan la forma más común de este tipo de enfermedades, que es el "scrapie" o prurito lumbar en ovejas y cabras.3,6 Los animales afectados pierden la coordinación motora hasta el punto en que les es imposible ponerse de pie, se tornan irritables y en algunos casos, sufren de un prurito intenso. Los síntomas por lo general se intensifican progresivamente y culminan con la muerte del animal en el transcurso de un año.6
Características de las enfermedades causadas por Priones
La enfermedad de Creutzfeldt–Jakob fue descrita por primera vez en 1920porH.G.CreutzfeldtyA. Jakob, yfue la primera EPRs reportada en humanos. En la mayoría de los casos, se presenta deforma esporádica;7,8 únicamente es hereditaria en 10 a 15% de los afectados, y han sido reportadas infecciones transmitidas portransplantes de córnea, implantes de duramadre, uso de electrodos cerebrales e instrumentos quirúrgicos contaminados asícomo por inyección de hormonas del crecimiento obtenidas de glándulas hipofisiarias humanas.3,6 La incidencia anual de la ECJ, que es la EPRs más frecuente, es de uno a dos casos por millón de personas.9,10 En 1936, J. Gerstmann, E. Stráusslere I. Scheinker describieron el síndrome que llevasus nombres. El síndrome de Gerstmann–Straussler–Scheinker es la única EPRs que se transmite exclusivamente deforma hereditaria, debido a mutaciones puntuales con carácter autosómico en el gen que codifica para la proteína prión. Se ha propuesto, que la sustitución de un aminoácido por otro en la secuencia de dicha proteína, altera su estabilidad termodinámica en el medio celular, lo cual favorece el cambio conformacional que le confiere su carácter patológico.3,11
El IFF es una enfermedad caracterizada por trastornos progresivos del sueño, y de los sistemas endocrino y muscular que culminan con la declinación cognoscitiva, pérdida de la independencia funcional y la muerte.9 La enfermedad fue reportada desde 1939 pero se describió por primera vez en 1986 en un individuo de 53 años de edad que se presentó con insomnio progresivo.3,10 E. Lugarest, R. Medori y P. Gambeti acuñaron el nombre del padecimiento refiriéndose al síntoma más característico y a que todos los casos reportados hasta entonces eran hereditarios. Sin embargo, recientemente ha sido identificada una EPRs de carácter esporádico con características clínicas y neuropatológicas muysimilaresalasdel IFF, aunque aún nose ha corroborado que se trate de la misma enfermedad.9,10 La AAC–PRP se distingue por presentar, además de la proteína prión anómala, lesiones neuronales ocasionadas por la proteína tau, similares a las que se presentan en la enfermedad de Alzheimer (EA). Consiste en un nuevo fenotipo de la enfermedad de GSS y se ha asociado también a mutaciones puntuales del gen que codifica para la proteína prión. Sin embargo, hasta el momento ha sido reportado un caso, lo que dificulta la realización de otros estudios al respecto.12
Vincent Zigas y Carleton Gajdusek describieron el kuru en 1957, es una EPRs endémica de los miembros de la tribu Fore de las islas de Papua de Nueva Guinea. Estudios epidemiológicos sugieren que el kuru era transmitido mediante la ingestión de los cadáveres de familiares en un ritual religioso, afectando mayoritariamente a mujeres y niños de ambos sexos que consumían el tejido cerebral.10,13 El cese del canibalismo en la década de 1950 propició la desaparición casi total de la enfermedad, hecho que evidencia el papel del endocanibalismo en la transmisibilidad del kuru. Los individuos afectados sufrían de cefalalgia y dolor articular, síntomas que precedían a la disfunción cerebelosa caracterizada por dificultad para caminar, pérdida de la coordinación motriz y temblores generalizados. La muerte ocurría en el transcurso de uno a dos años después de la presentación de los síntomas.10,14
En 1996, con el estudio de 10 casos de una nueva prionopatía, expertos del Ministerio de Salud del gobierno de Gran Bretaña informaron que el agente etiológico de la EEB ("enfermedad de las vacas locas") se había extendido a los humanos. El surgimiento de una nueva variante de la ECJ probablemente esté relacionado con la ingestión de tejido nervioso, muscular o de médula ósea de ganado infectado. Algunos trabajos experimentales han evidenciado la relación entre el agente causal de la EEB y la nvECJ, mediante la transmisión exitosa de la EEB a primates no humanos, encontrándose una estrecha similitud de sus perfiles neuropatológicos.1015 Las características clínicas de la nvECJ que difieren de la ECJ clásica incluyen: edad de inicio más temprana, duración más prolongada, cambios conductuales frecuentes y tempranos, así como la ausencia de las anormalidades electroencefalográficas típicas de la ECJ.9,10
Las EPRs presentan características neuropatológicas similares: pérdida neuronal, que le confiere al cerebro un aspecto espongiforme debido a los espacios que quedan en el tejido; formación anómala de gran cantidad de vacuolas intraneuronales de 20–200 µm de diámetro en cualquier capa de la corteza cerebral;16 presencia de cúmulos de células de la glía y de astrocitos que aumentan de tamaño como reacción al daño neuronal (astrogliosis);6 ausencia de reacción inflamatoria y de respuesta inmunológica y, finalmente, acumulación de depósitos de la proteína prión anómala.9,17 La presencia y magnitud de estas características varían en cada tipo de EPRs y en cada caso4,17 (Figura 1). Existe además una activación astrocítica abundante y daño pre–inflamatorio.58 Sin embargo, los datos histopatológicos no han sido consistentes.59 Evidencia reciente demuestra que la patología por priones tiene una respuesta inflamatoria altamente atípica, caracterizada por la activación de las poblaciones de macrófagos en el cerebro. Esto sugiere que una inflamación sistémica podría impactar en la inflamación local en el cerebro dañado y exacerbando la síntesis de citocinas inflamatorias u otros mediadores en el cerebro y que podrían contribuir al proceso crónico degenerativo.60
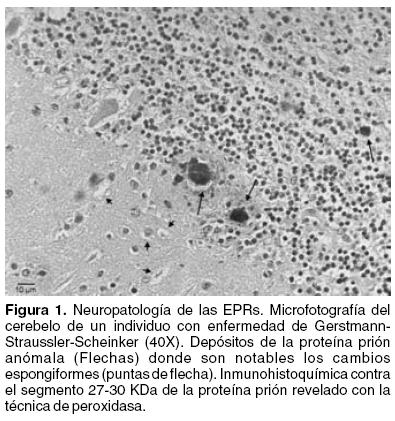
La proteína prión normal está distribuida de manera homogénea en todos los tejidos humanos. Sin embargo, en particular en la nvCJD, se ha mostrado que la acumulación anormal de la proteína se da, principalmente, en el tejido linfoide huésped, en particular en las células dendríticas foliculares.55 Cuando la neuroinvasión ocurre, se presenta por dos vías neuronales diferentes (vago y nervios esplénicos) y precede usualmente, a la propagación en órganos linfoides secundarios. Aunque otras formas etiológicas de la PrPSc (Proteína >> Prión Scrapie) aparentemente están limitadas al SNC.54 Así mismo, estudios longitudinales en modelos experimentales de scrapie, indican que los cambios neuropatológicos (activación glial y degeneración de las neuronas) están presentes antes de que aparezcan los síntomas y están topológicamente relacionados con el depósito de PrPSc.54 Sin embargo, queda abierta la interrogante de la aparición de la nvCJD, que en un principio se atribuyó al consumo de carne contaminada (vacas locas). Se ha sugerido, basándose en estudios con modelos animales, que la progresión de la infección al SNC es gradual, llegando hasta los niveles máximos durante la enfermedad. Por otra parte, reportes recientes sugieren que la nvCJD pudiera tener una transmisión iatrogénica. Ya sea por cirugía, transfusión sanguínea, o un transplante de los órganos de un donador en fase preclínica de una enfermedad por priones.55 Los datos más recientes al respecto indican, que las transfusiones sanguíneas pudieran ser el vínculo directo. Esta noticia ha causado revuelo en la comunidad médica y científica.56,57 Al menos dos casos se han reportado. En uno, el paciente murió por causas ajenas a una neurodegeneración.57 El paciente había recibido cinco años antes una transfusión sanguínea de un donador que desarrolló la nvCJD. La proteína prión se encontró en el primer paciente en varios órganos excepto en el cerebro (fase preclínica). La particularidad del caso fue que el paciente era además heterocigoto para el codón 129. Estos datos abren al menos dos posibilidades de prevención: una, se debe evitar y descartar como donadores de sangre a aquellas personas que hayan sido transfundidas alguna vez en su vida; y dos, que además de las condiciones de infección, existe una susceptibilidad genética para adquirir la nvCJD.57
La biología molecular de los priones
Utilizando técnicas de Biología Molecular se logró determinar la naturaleza del agente etiológico de las EPRs. Dado el carácter infeccioso de dicho agente, se supuso que este debía contener al menos un tipo de ácido nucleico. Según el dogma central de la Biología Molecular, de estas biomoléculas depende la única forma viable de reproducción. Sin embargo, al determinarse la carencia de ácidos nucleicos en su estructura, fue propuesto el término "prión", apócope de proteinaceous infectious particle para designar a la partícula cuyo único componente conocido como agente etiológico es una proteína.18,19 Se ha sugerido que las EPRs son causadas por la modificación post–traduccional de la Proteína Prion Celular (PrPc)—que es una proteína que se encuentra en las células neuronales de forma natural—, mediante un cambio de conformación de la estructura terciaria, resultando una isoforma patológica denominada PrPSc 20 La ausencia de respuesta inmune contra el prión y la presencia del gen que codifica para esta proteína en el genoma del hospedero, confirmaron que el agente causal es unaforma anormal de una proteína normal del organismo.6 Este descubrimiento se contrapone al principio biológico que establece que la secuencia de aminoácidos de una proteína determina de manera unívoca el plegamiento o estructura terciaria de esta, porque en la actualidad se conoce que existen al menos dos isoformas de la proteína prión cuyas estructuras son termodinámicamente estables como para mantenerse en condiciones fisiológicas.11 Además, no han sido encontradas diferencias químicas con excepción de los casos hereditarios, ni de estructura covalente entre ambas isoformas.21,22
La PrPc se codifica por un único gen llamado PRNP presente en los mamíferos, aves e incluso en los reptiles;1,23 la secuencia de aminoácidos se ha mantenido en la escala filogenética. Se presenta en tejido nervioso, muscular y en células del sistema inmunitario.24 La mayor concentración de PrPc ha sido reportada en las neuronas, particularmente en las membranas pre y post–sinápticas, lo que sugiere que posee importancia en el funcionamiento de éstas,23 sin embargo, su función específica no ha sido dilucidada aún.25 Mediante un proceso hasta el momento desconocido, la PrPc cambia su conformación espacial y se convierte en una forma anómala PrPSc que no cumple ninguna función en la célula y cuya presencia es causa de patología. Los mecanismos que intervienen en las manifestaciones clínicas y neuropatológicas de las EPRs tampoco son conocidos. Únicamente existe evidencia de que la PrPSc ejerce efectos tóxicos sobre las neuronas que pueden conducirlas a la muerte mediante apoptosis. La presencia de gran cantidad de vacuolas intraneuronales se debe a la división de la tricapa lipídica de las membranas neuronales, modificación probablemente relacionada con el hecho de que PrP es una proteína integral de dichas membranas.9 La acumulación de astrocitos reactivos y de células de la glía hipertróficas probablemente sea consecuencia del daño neuronal.6
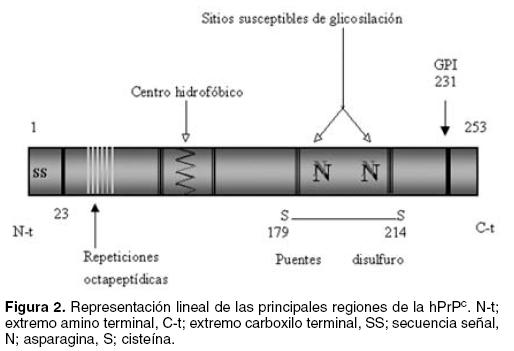
La proteína prión celular humana (hPrPc) –que consta de 253 aminoácidos— es sintetizada en los ribosomas del retículo endoplásmico (RE) y es translocada de manera cotraduccional al interior de este organelo debido a la acción de una secuencia señal constituida por los primeros 22 aminoácidos del extremo amino terminal (N–terminal) de la proteína.26 Una vez dentro del RE, el procesamiento postraduccional normal incluye la eliminación proteolítica de la secuencia señal (aminoácidos 1–22) y la eliminación de 22 aminoácidos de su extremo carboxilo terminal (C–terminal). Posteriormente, le son adicionadas una o dos cadenas de oligosacáridos conocidas como glicosilfosfatidilinositol (GPI), probablemente en la serina número 231,9,27 El GPI se une covalentemente a los lípidos de la membrana plasmática de las células de manera que la proteína queda expuesta al medio extracelular. Cerca del extremo N–terminal se encuentra una región que contiene 4 repeticiones del octapéptido PHGG(G/S)WGQ, rico en residuos prolina y glicina (entre los residuos 60 y 93) y una secuencia homologa que ha perdido un residuo de histidina PQGGGGWGQ (entre los residuos 52 y 60), posteriormente se encuentra una zona rica en aminoácidos hidrofóbicos. Existen dos sitios susceptibles de glicosilación, particularmente de N–glicosilación, en las Asparaginas 181 y 197. Además, los residuos 179 y 214 están unidos mediante un enlace disulfuro28 (Figura 2).
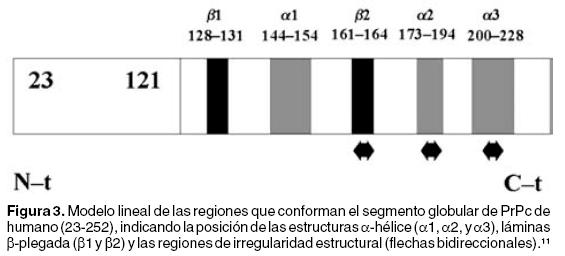
Estudios realizados mediante Resonancia Magnética Nuclear muestran que la hPrPc posee un dominio globular que se extiende desde el aminoácido 125 hasta el 228. En este dominio se forman tres estructuras α–hélice en los residuos 144–154, 173–194 y 200–228, así como dos dominios de láminas β–plegada de los residuos 128–131 y 161–164. Existen tres zonas de irregularidad estructural, la primera es un asa constituida por los aminoácidos 167–171, la segunda comprende a los aminoácidos 187–194 y la tercera, a los residuos 219–228 29 (Figura 3).
Pese a los avances logrados en el conocimiento de la forma celular de la proteína prión de varios organismos, la biología celular y la estructura de la forma patológica PrPSc aún se desconocen. Han logrado diferenciarse en la práctica debido a que ambas isoformas difieren notablemente en sus propiedades químicas y fisiológicas.6 La PrPc está compuesta por 42% de estructuras α–hélice y un 3% de láminas β–plegada. Es un monómero presente en la superficie celular, sensible a la digestión por proteasas y soluble en detergentes.30 Se ha determinado que la PrPSc está formada por un 43% de láminas β–plegada y 30% de α–hélice.30 Es una proteína que tiende a agregarse para formar oligómeros insolubles, es parcialmente resistente a la degradación por proteasas e insoluble en presencia de detergentes.31
Teorías del cambio conformacional de los priones
Evidencias experimentales han probado que el evento fundamental y probablemente, causal, de las EPRs es el cambio conformacional de la PrPc a la proteína prión anómala o "scrapie" PrPSc 32 por lo que, en la actualidad, la mayoría de las investigaciones sobre la etiología de las EPRs se han centrado en la elucidación de este fenómeno. La conversión de PrPc en PrPSc es un evento postraduccional que ocurre después de que la proteína "normal" PrPc ha alcanzado su posición en el dominio extracelular de las membranas neuronales, o incluso, mucho después, durante el transporte vesicular de la proteína al interior de la neurona.1 Debido a la existencia de distintos mecanismos de adquisición de las EPRs –hereditario, esporádico e infeccioso– se han propuesto varias teorías que pretenden explicar el cambio conformacional, y se mencionan a continuación.
Desestabilización de la PrPC debida a mutaciones del gen PRNP
Las variantes hereditarias de las EPRs se deben a mutaciones puntuales del gen PRNP que se manifiestan en la sustitución de un aminoácido por otro en la PrP,33 o por la inserción de repeticiones de aminoácidos en múltiplos de ocho.3,34 Stanley Prusiner sugiere que la presencia de aminoácidos incorrectos podría desestabilizar la estructura terciaria de la proteína, principalmente, en los dominios α–hélice, aumentando la posibilidad de que la hélice afectada y sus vecinas se plieguen nuevamente constituyendo estructuras en lámina β–plegada.6 En la actualidad, están caracterizadas alrededor de 30 mutaciones en familias con EPRs heredadas,10 sin embargo, únicamente 10% de los casos reportados de EPRs son de origen hereditario, por lo que la vasta mayoría son esporádicas e infecciosas y no presentan algún tipo de mutación en el gen, ni alteraciones en la secuencia de aminoácidos de la proteína.6 Por esta razón, esta teoría no está lo suficientemente argumentada como para explicar el cambio conformacional de 90% de los casos.11 Para probar la validez de esta teoría en las EPRs hereditarias, se ha estudiado la estabilidad termodinámica de ocho diferentes PrPc cuya secuencia difiere únicamente en un aminoácido. Se ha demostrado que estas mutaciones puntuales están asociadas a EPRs, pero se demostró que ningún cambio de amino ácidos causa la inestabilidad suficiente para provocar el cambio de conformación característico de la PrPSc, por lo que se sugiere que la desestabilización de la PrPc no es el mecanismo causal de la formación de la isoforma patológica en las variantes hereditarias.11
Interacciones moleculares entre PrPC y PrPSc
Esta teoría incluye dos modelos de explicación: El primero se denomina Modelo de Nucleación. Sugiere que la isoforma "normal" PrPc se encuentra en equilibrio conformacional con la isoforma patológica PrPSc o con un precursor de esta. Aunque la estructura de PrPc es favorecida, el cambio conformacional es un proceso estocástico que ocurre cuando unas cuantas moléculas de conformación anormal actúan como una semilla o núcleo capaz de inducir el cambio masivo de moléculas normales en moléculas con la conformación anómala19,34 y es el resultado de interacciones directas entre la PrPc y la PrPSc.18 Una vez que se inicia la transformación, ésta se propaga como una reacción en cadena, y debido a la insolubilidad que caracteriza a las moléculas de PrPSc, éstas se depositan en el citoplasma neuronal formando extensos agregados que dañan a las células.19,30 El proceso se asemeja al fenómeno de nucleación de los cristales, en el que una vez presente el cristal "semilla", la agregación es ampliamente favorecida y ocurre de manera relativamente rápida.1 Estudios realizados por Come y cois, y por Glover y cois., en péptidos basados en secuencias de proteínas prión y en el factor de la levadura [PSI+] respectivamente, han demostrado que el mecanismo de conversión de los priones y la formación de agregados es dependiente del proceso de nucleación16,35,36 (Figura 4).
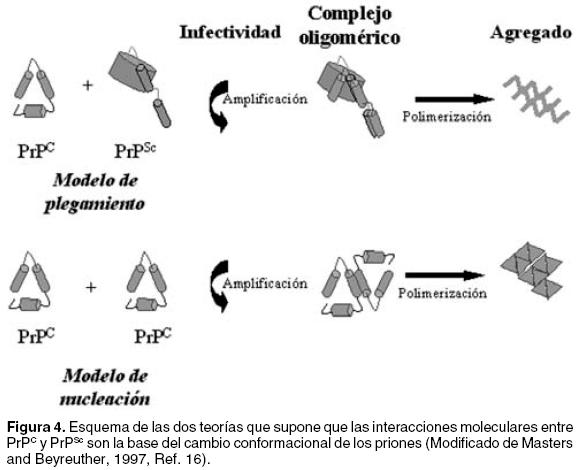
El segundo, Modelo de Plegamiento, propone que la conversión de la PrPc requiere que esta se encuentre desplegada y que se vuelva a plegar de manera anormal bajo la influencia de una molécula de PrPSc.37 Este proceso requiere franquear una barrera energética muy grande.1,38 La presencia de la PrPSc que iniciaría este proceso sería debida a la infección o a la transformación esporádica de moléculas PrPc.39 Este modelo ha sido apoyado al reportarse que en estudios in vitro la PrPc, conformada mayoritariamente por estructuras α–hélice, es capaz de cambiar espontáneamente de conformación hacia estructuras β–plegada que constituyen la PrPSc 9,40 (Figura 4). Como implicación inmediata de estos dos modelos de explicación del cambio conformacional, se intuye que es necesaria la presencia de la PrPc en el hospedero para que pueda ser establecida una infección; esto fue corroborado por Prusiner en 1982, al demostrar la resistencia de ratones con el gen PRNP suprimido, y que por lo tanto, no producían PrPSc al ser infectados por scrapie.6
Procesos postraduccionales
Debido a que no se han encontrado diferencias químicas entre la PrPc y la PrPSc, se presume la existencia de otros procesos implicados en el cambio conformacional y en la agregación de la PrP. Esto permite explicar la patología de las prionopatías a través de las modificaciones postraduccionales de la PrP. Un proceso similar se estudia ya en la EA, donde se ha observado la importancia que tienen los procesos postraduccionales de las proteínas involucradas que se agregan y se vuelven insolubles, la proteína tau y amiloide–Β.41,42 Al parecer, tanto en la EA y en las enfermedades por priones existen mecanismos comunes en la agregación e insolubilidad de las proteínas. Una proteína no es biológicamente activa hasta que adquiere la conformación plegada nativa, determinada por su secuencia de aminoácidos. La cadena polipeptídica adopta espontáneamente, durante o después de su síntesis, dicha conformación. De este modo, el mensaje genético lineal del que es portador el ARN mensajero, se convierte en una estructura tridimensional específica del nuevo polipéptido sintetizado. Sin embargo, en muchos casos es necesario que la cadena polipeptídica —recién sintetizada— experimente una modificación pos–traduccional. Finalmente, la proteína adopta su conformación nativa respectiva. A estos cambios, se les denomina modificaciones postraduccionales y dependen de la naturaleza de la proteína. Uno de los mecanismos postraduccionales de mayor importancia en la fisiopatogénesis de las EETs, es la glicosilación.
Glicosilación de la PRPC
La glicosilación consiste en la adición covalente de oligosacáridos a las proteínas y lípidos de manera enzimáticay donde los carbohidratos quedan como cadenas laterales. Las proteínas glicosiladas o glicoproteínas tienen funciones importantes en membranas, lisosomas y en el espacio extracelular, participando principalmente como moléculas de reconocimiento celular. En contraste, pocas proteínas del citosol se encuentran glicosiladas.
Las glicoproteínas contienen una o más cadenas de carbohidratos, que están clasificadas de acuerdo con el aminoácido al que se une el azúcar; tipo N–glicosídico si se enlaza sobre una asparagina (Asp), y del tipo O–glicosídico cuando la transferencia se realiza con serina (Ser) o treonina (Thr).
La PrPc posee dos sitios propicios para cadenas de N–oligosacáridos (en los residuos 180 y 196) y en el C–terminal (residuo 179). Los sitios son ocupados en forma variable, conformando cuatro glicoformas de la proteína, mismas que coexisten. Una está doblemente glicosilada, dos monoglicosiladas y una no glicosilada.43 Los oligosacáridos que están unidos a la PrPc, son N–oligosacáridos, establecen formas ortogonales, con carga negativa. Éstos cubren a la proteína impidiendo, por efecto estérico, las interacciones intramoleculares o intermoleculares. El hecho que la PrP contenga tal variedad de complejos oligosacarídicos, sugiere que se modifican las propiedades que distinguen entre la PrPc y PrPSc. Sin embargo, células tratadas con un inhibidor de la glicosilación, como la tunicamicina, produjeron especies simples de la PrP no glicosiladas, pero resistentes a proteasas. En este experimento se concluyó que la N–glicosilación no es esencial para la síntesis de la PrP resistente a proteasas. Sin embargo, no hay evidencia de que las moléculas de PrPSc no glicosiladas se asocien a la infectividad en las enfermedades por priones. Por otra parte, los modelos propuestos de la PrPc, indican sitios probables de unión para la proteína X. Esto indica que la presencia de las cadenas de carbohidratos no constituye un impedimento para la asociación entre la PrPc y la proteína X teórica propuesta. Aunque en estos modelos aparece un número importante de sitios posibles para la O–glicosilación, no se han detectado O–glicanos en la PrPc de hámster. Sin embargo, nuestro grupo ha demostrado la existencia de residuos O–glicosilados en los depósitos de PrPSc en el síndrome de GSS.17 Hasta el momento se ha sugerido que las modificaciones postraduccionales que pudieran dar lugar al cambio conformacional de PrPc a PrPScson producto de la N–glicosilación.44–46 La O–glicosilación no ha sido tomada en cuenta para explicar la trasformación de la PrP, ya que en las fracciones aisladas, tanto de PrPccomo de PrPsc, no se han encontrado este tipo de cadenas de carbohidratos. Por la dificultad para purificar fracciones de ambas isoformas de la PrP, y por la degradación, es factible suponer que los carbohidratos O–glicosilados pudieran estar presentes en la proteína prión y que no hayan sido detectados.46 Chen y cois, evaluaron los efectos que puede tener la adición de carbohidratos O–glicosilados al fragmento 108–144 de la proteína prión. Observaron que la adición de α–GalNAc en la Serina 135 evita la formación de agregados fibrilares, no así el carbohidrato β–GIcNAc que incluso potencia la agregación fibrilar del péptido, al igual que el α–GalNAc pero en la serina 132.46
Posibles funciones de los priones
Las funciones biológicas de PrPc permanecen poco claras hasta ahora. Sin embargo, dado que su secuencia aminoacídica es altamente conservada entre especies, se sugiere que posee especial importancia en procesos fisiológicos. Se ha reportado que PrPc liga iones de cobrey que este estimula la endocitosis de la PrPc desde la superficie celular de manera rápida e irreversible.47,48 Esto abre la posibilidad de que la PrPc funcione como receptor celular de Cu++ 23. Se ha propuesto también que PrPc actúa como transductor de señales intracelulares para la protección neuronal, en este proceso de señalización están involucradas moléculas como el adenosín monofosfato cíclico (cAMP) y fosfocinasas. La señal generada estuvo ausente en ratones carentes del gen PRNP.49 Otros estudios sugieren que la proteína prión está involucrada en el fenómeno de potenciación a largo plazo, en procesos fisiológicos del sueño y en mecanismos de adhesión célula–matriz extracelular.49–51
Alternativas terapéuticas experimentales
Durante los últimos 30 años se han usado más de 60 compuestos diferentes para tratar animales infectados experimentalmente. La especulación actual acerca de la importancia de la estructura química que pueda tener tanto una fase soluble en agua y una lipídica, podría tener aplicación directa para impedir el ensamble erróneo de la proteína prión en la membrana celular.52 Los compuestos que reducen la acumulación de PrPSc en modelos celulares en cultivos infectados, incluyen compuestos polianiónicos, rojo Congo, anfotericina B, porfirinas y derivados de fenotiazidas como la quinacrina.53 Sin embargo, estas drogas han demostrado ser útiles solamente en animales infectados periféricamente (después de la infección intraperitoneal) y antes de la invasión neuronal. Hay dos excepciones, el Polisulfatode Pentosany laanfotericinaque inyectados intraventricularmente en ratones que fueron infectados intracerebralmente, retardan el inicio clínico cuando se administran en el curso tardío de la infección.53 Algunos tratamientos inmunológicos se han probado de igual forma. Se ha tratado a los animales de experimentación con inmunidad pasiva y activa. Ejemplos concretos han demostrado que el uso de ciertos epítopes de PrP inhibe la propagación de PrPSc en cultivos celulares. En ratones transgénicos con el gen PRNP, se ha logrado una protección periférica, pero no central, contra la infección por priones. Algunos otros intentos se han realizado usando ingeniería genética, han sido alentadores y podrían neutralizar la infección in vivo.53 Sin embargo, los compuestos más efectivos contra la infección, probados hasta el momento, deben ser administrados al mismo tiempo que la infección se produce o en un plazo inmediato. Por lo que estas patologías siguen siendo incurables hasta el momento.54
Conclusión
Las enfermedades causadas por priones son, por múltiples razones, patologías sin precedente. Son enfermedades neurodegenerativas de carácter esporádico, infeccioso y hereditario; el mecanismo de transmisión de éstas contradice el Dogma Central de la Biología Molecular ya que el agente etiológico es capaz de replicarse a sí mismo en ausencia de ácido nucleico, y además, viola el principio biológico que establece que la secuencia de aminoácidos de una proteína determina de manera unívoca el plegamiento o estructura terciaria de ésta. La generación de estos nuevos conocimientos fue motivo del otorgamiento de dos premios Nobel; al doctor Carleton Gajdusek en 1976 por haber demostrado la transmisibilidad del kuru, y al doctor Stanley Prusiner en 1997 por descubrir la naturaleza química del agente causal de estas enfermedades. Sin embargo, el evento clave en el desarrollo de las EPRs, el cambio conformacional de la proteína prión, permanece en el ámbito de las hipótesis. De esta manera, se pone de manifiesto la necesidad de involucrar esfuerzos para la generación y rectificación de conocimiento concerniente a este fenómeno.
Proyecto apoyado parcialmente por CONACYT MO–334; FUNSLUD y PAPIIT–UNAM.
Referencias
1. Weissmann C. Molecular genetics of Transmisible Sponglform Encephalopathies. J Biol Chem 1999; 274;1:3–6. [ Links ]
2. Weissmann C. The state of the prion. Nat Rev Microbiol 2004; 11:861–871. [ Links ]
3. Belay ED. Transmissible spongiform encephalopathies in humans. Ann Rev Microbiol 1999; 53:283–314. [ Links ]
4. Bendheim PE, Bockman MP, McKinley MP, Kingsbury DT, Prusiner S. Scraple and Creutzfedtl–Jakob disease prion proteins share physical properties and antigenic determinants. Proc Nati Acad Sel USA 1985; 82:997–1001. [ Links ]
5. Kovacs GG, Kalev O, Budka H. Contribution of neuropathology to the understanding of human prion disease. Folia Neuropathol 2004;42 Suppl A: 69–76. [ Links ]
6. Prusiner S. Novel Protelnaceous Infectious Particles cause Scraple. Science 1982;216:136–144. [ Links ]
7. Mallucci G, Collinge J. Update on Creutzfeldt–Jakob disease. Curr Opin Neurol 2004;6:641–647. [ Links ]
8. Croes EA. Therapeutic approaches In treating Creutzfeldt–Jakob disease –what does the future hold? Expert Opin Pharmacother 2004; 11:2391–2396. [ Links ]
9. Coria BF. Demencias por priones. En: Alberca R, López Pousa YS. Enfermedad de Alzheimer y otras demencias. Médica Panamericana. España. 2002. [ Links ]
10. Weihl CC, Ross RP. Enfermedad de Creutzdeldt–Jakob, nueva variedad de la enfermedad de Creutzfeldt–Jakob, y la encefalopatía espongiforme bovina. En: Clínicas Neurológlcas de Norteamérica. Infecciones del Sistema Nervioso Central. Vol. 4. McGraw–Hill Interamerlcana. México 1998. [ Links ]
11. Liemann S,.GIockshuber R. Influence of amlno add substitutions related to inherited prion disease on the thermodynamlc stability of the cellular prion protein. Blochem 1999;38:3258–3267. [ Links ]
12. Kitamoto T, Lizuka R, Tateishi YJ. An amber mutation of prion protein In Gerstmann–Straussler–Schelnker syndrome with mutant PrP plaques. Biochem Biophys Res Commu 1993; 192:525–531. [ Links ]
13. Liberski PP, Gajdusek DC. Kuru: Forty years later, a historical note. Brain Pathol 1997; 7:555–560. [ Links ]
14. Hainfellner JA, Liberski PP, Guiroy DC, Cervenakova L, Brown P, Gajdusek DC, et al. Pathology and Inmunocytochemistry of a Kuru Brain. Brain Pathol 1997; 7:547–553. [ Links ]
15. Aguzzi A, Bitifler T, Klein M, Brandner S, Raeber A, Flechsing E, Weissmann C. Neurotoxlclty and Neuroinvasiveness of prions. Brain Pathol 1997; 7:1137–1138. [ Links ]
16. Masters CL, Beyreuther K. Tracking turncoat prion proteins. News and Views. Nature 1997;388:228–229. [ Links ]
17. Sánchez A, Guzman A, Ortiz A, Rembao D, Espinosa B, Zenteno E, Guevara J. Toluidine blue–0 staining of prion protein deposits. Hlstochem Cell Biol 2001;116:519–524. [ Links ]
18. Prusiner S. El prion en la patología. Investigación y Ciencia. 1995; Marzo: 14–21. [ Links ]
19. Aranda A. Possible cell–free prion replication. Med Hypotheses 1992;38:249–251. [ Links ]
20. Tatzelt J, Winklhofer KF. Folding and misfolding of the prion protein in the secretory pathway. Amyloid 2004; 3:162–172. [ Links ]
21. Zhang H, Stockel J, Mehlhorn I, Groth D, Baldwin MA, Prusiner S, Lames TL, Cohen F. Physical studies of conformatlonal plasticity In a recomblnant prion protein. Blochem 1997;36:3543–3553. [ Links ]
22. Kuwata K, Li H, Yamada H, Legname G, Prusiner S, Akasaka K, James T. Locally disordered conformer of the hamster prion protein: A crucial Intermediate to PrPSc? Blochem 2002; 41:12277–12283. [ Links ]
23. Brown DR. Prion and prejuice: normal protein and the synapse. Trends of Neurosclence 2001; 24:85–90. [ Links ]
24. Horiuchi M, Yamazaki N, Ikeda T, Ishiguro N, Shinagawa M. A cellular form of prion protein (PrPC) exists in many non–neuronal tissues of sheep. J Gen Virol 1995; 76:2583–2587. [ Links ]
25. Brockes J. Topics in prion cell biology. Current Opinion in Neurobiol 1999; 9:571–577. [ Links ]
26. Hölscher C, Bach UC, Dobberstein B. Prion Protein Contains a Second Endoplasmic Reticulum Targeting Signal Sequence Located at its C Terminus. J Biol Chem 2001; 276:1 3388–1 3394. [ Links ]
27. Stahl N, Baldwin MA, Burlingame A, Prusiner S. Identification of Glycolnosltol phospholipid linked and truncated forms of the scraple prion protein. Blochem 1990;29:8879–8 [ Links ]
28. Nandi PK, Leclerc E, Marc D. Unusual property of prion protein unfolding in neutral salt solution. Biochem 2002; 41:11017–11024. [ Links ]
29. Zahn R, Liu A, Liihrs T, Riek R, Von Schroetter C, López F, Billeter M, Calzolai L, Wider G, Wüthrich K. NMR solution structure of human prion protein. Proc Nati Acad Sci USA 1999; 97:145–150. [ Links ]
30. Pan KM, Baldwin MA, Nguyen J, Gassey M, Serban A. Conversion of α–hellces into β–sheets features in the formation of the scraple prion proteins. Proc Nati Acad Sci USA 1993; 90:10962–10966. [ Links ]
31. Bell JE, Ironside JW. Neuropathology of spongiform encephalopathies in humans. Brain Med Bull 1993; 49:738–777. [ Links ]
32. DebBurman S, Raymond G, Caughey B, Lindquist A. Chaperone–supervised conversion of prion protein to its protease–resistant form. Proc Nati Acad Sci USA 1997; 94:13938–13943. [ Links ]
33. Cohen FE. Protein misfolding and prion diseases. J Mol Blol 1999; 293:313–320. [ Links ]
34. Fernandez–Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence–dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 2004; 10:1302–1306. [ Links ]
35. Castilla J, Hetz C, Soto C. Molecular mechanisms of neurotoxicity of pathological prion protein. Curr Mol Med 2004; 4:397–403. [ Links ]
36. Jarrett J, Lansbury PT. Seeding "One–Dimensional crystallization" of amyloid A pathogenic mechanism in Alzheimer's disease and Scraple? Cell 1993;73:1055–1058. [ Links ]
37. Georgieva D, Koker M, Redecke L, Perbandt M, Clos J, Bredehorst R, Genov N, Betzel C. Oligomerization of the proteolytic products is an intrinsic property of prion proteins. Biochem Biophys Res Commun 2004; 323:1278–1286. [ Links ]
38. Gasset M. El prion, una herejía científica en la vida cotidiana. Fronteras de la Ciencia y Tecnología (Francia) 1996;13:4–6. [ Links ]
39. Satheeshkumar KS, Murali J, Jayakumar R. Assemblages of prion fragments: novel model systems for understanding amyloid toxicity. J Struct Blol 2004; 148:176–193. [ Links ]
40. Kocisko A, Come H, Priola A. Cell free formation of protease resistant prion protein. Nature 1994; 370:471–474. [ Links ]
41. Guevara J, Espinosa B, Zenteno E, Vázquez L, Luna J, Perry G, Mena R. Altered glycosylation pattern of proteins in Alzheimer Disease. J. Neuropathol Exp Neurol 1998; 57:905–914. [ Links ]
42. Espinosa B, Guevara J, Hernández P, Slomianny MC, Guzman A, Martinez–Cairo S, Zenteno E. Characterization of an O–glycosylated plaque–associated protein from Alzheimer's disease brain. J Neuropathol Exp Neurol 2003; 62:34–41. [ Links ]
43. Clausen H, Benneth EP. One family de UDP–GalNAc: polypeptlde N–acetyl–galactosaminyltransferases control the initiation of mucin–type O–linked glycosylation. Glycobiology 1996; 6:635–646. [ Links ]
44. Rudd PM, Endo T, Colominas C, Groth D, Wheeler SF, Harvey DJ, et al. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc Nati Acad Sci USA 1999; 96:13044–13049. [ Links ]
45. Rudd PM, Wormald MR, Wing DR, Prusiner SB and Dwek RA. prion glycoprotein: Structure, dynamics and roles for sugars. Biochemistry 2001; 40:3759–3766. [ Links ]
46. Chen PY, Lin CC, Chang YT, Lin SC, Chan SI. One O–linked sugar can affect the coil–to–beta structural transition of the prion peptide. Proc Nati Acad Sci USA 2002; 99:12633–12638. [ Links ]
47. Quaglio E, Chiesa R, Harris A. Copper converts the Cellular prion protein Into a protease–reslstant species than Is distinct from the scraple isoform. J Blol Chem 2001; 276:11432–11438. [ Links ]
48. Kuczius T, Buschmann A, Zhang W, Karch H, Becker K, Peters G, Groschup MH. Cellular prion protein acquires resistance to proteolytic degradation following copper ion binding. Biol Chem 2004;385:739–747. [ Links ]
49. Martins VR, Brentani RR. The biology of the cellular prion protein. Neurochem International 2002; 41:353–355. [ Links ]
50. Dormont D. Prion diseases: pathogenesis and public health concerns. FEBS Letters 2002; 529:17–21. [ Links ]
51. Cashman NR, Caughey B. Prion diseases, close to effective therapy?. Nat Rev Drug Discov 2004; 10:874–884. [ Links ]
52. Brown P. Drug therapy in human and experimental transmissible sponglform encephalopathy. Neurology 2002;58:1720–1725. [ Links ]
53. Mallucci G, Collinge J. Rational targeting for prion therapeutics. Nat Revs 2005;6:23–34. [ Links ]
54. Rossi G, Salmona M, Forloni G, Bugiani O, Tagliavini F. Therapeutic approaches to prion diseases. Clin Lab Med 2003;23:187–208. [ Links ]
55. Ramasamy I, Law M, Collins S, Brooke F. Organ distribution of prion proteins in variant Creutzfeldt–Jakob disease. Lancet Infect Dis 2003; 3:214–222. [ Links ]
56. Hopkin M. Fears grow as blood stocks pass on prion undetected. Nature 2004; 430:712. [ Links ]
57. Peden AH, Head MW, Ritchie Dl, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004; 264:527–529. [ Links ]
58. Van Everbroeck B, Dewulf E, Pals P, Lubke U, Martin JJ, Cras P. The role of cytoklnes, astrocytes, microglia and apoptosis in Creutzfeldt–Jakob disease. Neurobiol Aging. 2002; 23:59–64. [ Links ]
59. Perry VH, Cunningham C, Boche D. Atypical inflammation in the central nervous system in prion disease. Curr Opin Neurol. 2002; 15:349–354. [ Links ]
60. Perry VH. The Influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004; 18:407–413. [ Links ]

