










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta médica de México
versión On-line ISSN 2696-1288versión impresa ISSN 0016-3813
Gac. Méd. Méx vol.142 no.2 Ciudad de México mar./abr. 2006
Ejercicio clínico patológico
Hombre de 35 años con úlceras recurrentes en boca y regiones periuretral y escrotal
A 35–year– old man with recurrent mouth, periuretral, and scrotal ulcers
Norma E. Rivera–Martínez,ª* Haiko Nellen–Hummel,ª Andrés Jacobo–Ruvalcaba,ª Lucero Kameyama–Fernández,ª Gisela N. Hernández–Luis,ª Enrique Blanco,b Jesús Medrano,c José Halabe–Cheremª
Departamentos de ª Medicina Interna,
b Patología y
c Hematología.
Hospital de Especialidades del Centro Médico Nacional Siglo XXI, IMSS México D. F., México
*Correspondencia y solicitud de sobretiros:
Dra. Norma E. Rivera–Martínez,
Departamento de Medicina Interna, Hospital de Especialidades, CMN SXXI, IMSS.
Av. Cuauhtémoc No. 330, Col. Doctores, Del. Cuauhtémoc,
México D. F. C. P. 06725.
Coordinador: Dr. Manuel de la Llata–Romero
Colaboradores: Dr. Juan Urrusti–Sanz
Dr. Jesús Aguirre–García
Dr. Roberto Sánchez–Ramírez
Presentación del caso
Hombre de 35 años de edad, con historia de aparición de úlceras pequeñas dolorosas, localizadas en cavidad bucal, región periuretral y escrotal desde hace 4 años, de carácter recurrente. Ingresó al hospital por padecimiento de 6 meses de evolución caracterizado por pérdida de peso, fiebre sin predominio de horario, artralgias en codos, muñecas, articulaciones metacarpofalángicas, rodillas y tobillos. Se automedicó con antiinflamatorios no esteroideos, con uso indiscriminado, sin remitir la sintomatología. Posteriormente aparece aumento de volumen en articulaciones metacarpofalángicas y rodillas, dolor y parestesias en miembros pélvicos con limitación de la deambulación, equimosis palpebral y hemorragia conjuntival bilateral, aparición de una dermatosis diseminada en tronco y extremidades, caracterizada por pápulas violáceas de 0.5 cm de diamétro con bordes bien definidos en cara anterior de piernas, aparecieron nódulos subcutáneos de medio centímetro de diámetro, no dolorosos. Al examen físico se identificaron úlceras en paladar blando y duro. En el fondo de ojo se identificó hemorragia retiniana y la existencia de exudados blancos compatibles con vasculitis retiniana. La prueba de patergia fue negativa.
Los anticuerpos antinucleares, antivirus de la inmunodeficiencia humana y antivirus de la hepatitis B fueron negativos, al igual que el factor reumatoide, los hemocultivos, mielocultivos y urocultivos. La biometría hemática mostró anemia persistente y la química sanguínea, los electrólitos séricos y el examen general de orina no mostraron alteraciones. La radiografía de tórax y senos paranasales, así como el utltrasonido y tomograf ía de abdomen, el electrocardiograma y el ecocardiograma mostraron parámetros dentro de la normalidad. La electromiografía detectó cambios que fueron sugestivos de neuropatía axonal de todas las extremidades. El examen de médula ósea identificó cambios dispoyéticos en la serie mieloide, así como detención en la maduración, linfocitos granulares e hiperplasia de megacariocitos. En la biopsia de piel se identificó vasculitis linfocítica de vénulas postcapilares y trombosis in situ. El cariotipo fue 46 XY (10/ mo) con hipodiploidia en menos de 36 cromosomas.
Basados en la presencia de úlceras bucales y genitales, neuropatía periférica, vasculitis linfocítica en piel y retina, así como en los criterios diagnósticos del Colegio Americano de Reumatología, se diagnosticó enfermedad de Behçet. Por el hallazgo de vasculitis retiniana, se decidió iniciar tratamiento con prednisona a dosis de 1 mg/kg/día, con respuesta al tratamiento satisfactoria, con remisión de úlceras bucales, lesiones en ojos, piel y articulaciones. El síndrome mielodisplásico secundario que presentó el paciente fue clasificado como anemia sideroblástica refractaria a tratamiento.
Discusión clínica
La enfermedad de Behçet y el síndrome mielodisplásico es una asociación rara y debido al riesgo de desarrollar leucemia mielomonocítica debe vigilarse su evolución. Nuestro paciente tenia anemia refractaria al tratamiento, (que posteriormente fue clasificada como anemia sideroblástica) y datos compatibles con los criterios diagnósticos de enfermedad de Behçet.
La enfermedad de Behçet es una vasculitis de pequeños y grandes vasos de causa desconocida, afecta a capilares y vénulas, su prevalencia es difícil de determinar ya que muchos casos no son diagnosticados. Esta vasculitis es frecuente en el extremo y medio oriente. En Ankara, Turquía, la prevalencia es del 0.11% aproximadamente.1 En México se desconoce la prevalencia de la enfermedad y nunca se ha reportado un caso asociado a síndrome mielodisplásico. El alelo HLA–B51 se asocia frecuentemente a la enfermedad de Behçet, se relaciona con la severidad y es más común en pacientes con uveítis posterior y afección del sistema nervioso central. Los autoanticuerpos no forman parte del espectro de la enfermedad, sin embargo se han identificado anticuerpos contra las células endoteliales dirigidos contra la α–enolasa de la pared vascular, se han propuesto dichos anticuerpos como marcadores del Síndrome de Behçet pero aún no han sido aceptados. Las células T γδ han sido asociadas con la uveítis que se presenta en algunos pacientes; esta ha sido la anormalidad inmunológica más frecuente encontrada en Behçet.1
La enfermedad de Behçet no tiene signos y síntomas patognomónicos, ni características de laboratorio específicas. El diagnóstico se realiza con los criterios propuestos por el grupo de Estudio Internacional para la Enfermedad de Behçet. Una persona tiene la enfermedad si presenta úlceras bucales recurrentes y dos de las siguientes lesiones: úlceras genitales recurrentes, lesiones oculares, lesiones dermatológicas y la prueba de patergia positiva. El diagnóstico diferencial incluye aftosis oral crónica, infección por virus herpes simple, Síndrome de Sweet y síndromes relacionados a HLA B27 tales como espondilitis anquilosante.2
En el caso presentado, el diagnóstico se efectuó con base en la historia de úlceras bucales recurrentes, y en las lesiones oculares y dermatológicas (Figuras 1, 2 y 3). Las úlceras bucales aparecen en 97 a 100% de los pacientes. Con frecuencia representan el síntoma principal y preceden por años al resto de las manifestaciones. Aparecen en lengua, paladar duro y blando, región gingival, mucosas bucal y labial, tienen el borde eritematoso y en la superficie se encuentra una seudomembrana de color amarillo, son dolorosas, las lesiones tienden a autolimitarse en 10 días. Otras manifestaciones gastrointestinales que se presentan en el 0 a 25% de los pacientes, son úlceras en íleon terminal y colon ascendente, frecuentes en los pacientes japoneses pero raras en otras áreas geográficas.5

El involucramiento gastrointestinal causa dolor abdominal, diarrea, melena y algunas veces perforación, siendo la región mas afectada la ileocecal. Histológicamente las ulceras de los pacientes con Behçet son indistinguibles de las ulceras de la enfermedad de Crohn, sin embargo el hallazgo de granulomas puede excuir la primera. Además, la prueba de patergia es negativa en los pacientes con enfermedad inflamatoria intestinal.2
Las úlceras genitales se identifican en 80–97%. Ocurren en el escroto, pene o vulva, son morfológicamente similares a las úlceras bucales, pero más profundas y con márgenes irregulares, también son dolorosas. Otras manifestaciones que suelen subdiagnosticarse son la ependidimitis (6%) las úlceras extragenitales que dejan cicatrices y presentan vasculitis en la biopsia y las pápulas de pseudoSweet.
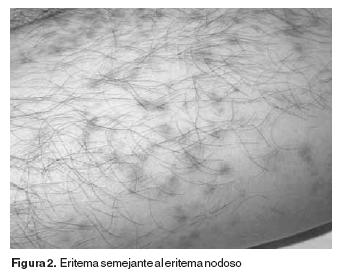
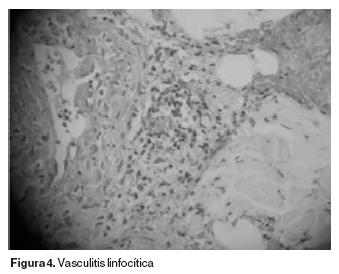
Las lesiones de piel se presentan en 69–80% de los casos. Las lesiones comprenden: seudofoliculitis, eritema nodoso, exantema acneiforme, lesiones pápulo pustulosas y raramente vasculitis manifestada como púrpura palpable.10 El eritema nodoso es común en los pacientes femeninos y usualmente ocurre en la región frontal de las piernas, las lesiones son dolorosas y se resuelve espontáneamente dejando un área pigmentada, algunas veces se ulceran (Figura 2). La seudofoliculitis y los nódulos acneiformes son más comunes en hombres, se distribuyen en la espalda, cara y cuello.2 Las manifestaciones cutáneas vasculíticas son caracterizadas por eritema nodoso "like", púrpura palpable, ampollas hemorrágicas, Síndrome Sweet "like", lesiones papulopustulosas sus características histológicas incluyen vasculitis leucocitoclástica en 17 a 42% de pacientes y vasculitis linfocítica en 31 a 42%. Las vasculitis cutáneas son predominantemente venulitis o flebitis en 48% de los pacientes, sugiriendo que la vasculitis asociada a enfermedad de Behçet debe separarse de otros tipos de dermatosis neutrof ílicas, ya que en buen número de casos, la vasculitis linfocítica es predominante13 (Figura 4). El eritema nodoso "like" observado en los pacientes con enfermedad de Behçet difiere del eritema nodoso clásico en sus características histopatológicas, ya que en Behçet está caracterizado por paniculitis lobular, septal o mixta, con adipocitos necróticos y vasculitis linfocítica, mientras que en el eritema nodoso clásico no se observa vasculitis.14,15
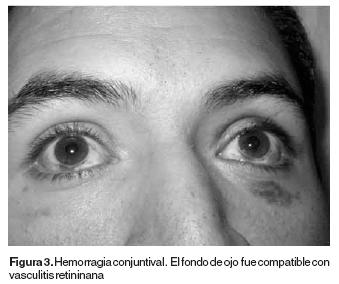
Las manifestaciones oculares son comunes en el curso de la enfermedad, en 50% de pacientes pueden identificarse. Los pacientes con lesiones oculares tienen usualmente algunos síntomas, que incluyen visión borrosa, dolor ocular, fotofobia, lagrimeo, hemorragia conjuntival, cuerpos fototes e hiperemia periglobal (como resultado de episcleritis) La uveítis anterior, con o sin hipopión (capa de pus en la cámara anterior) es el hallazgo más frecuente y es característica de la enfermedad de Behçet, se presenta de manera intermitente, cede espontáneamente pero produce cambios estructurales, causando deformidad del iris y glaucoma secundario. La alteración más grave es la vasculitis retiniana, que puede afectar arterias y venas y producir oclusión arterial, necrosis, neovascularización, desprendimiento de retina y hemorragia vítrea. La afección es frecuentemente bilateral. El resultado final de la vasculitits retiniana es un ojo ciego y doloroso. Otras alteraciones menos frecuentes son: glaucoma de ángulo cerrado, conjuntivitis, queratitis, escleritis, parálisis de nervios craneales, papiledema y neuropatía óptica isquémica. La sarcoidosis y la retinitis viral, algunas veces, tienen características indistinguibles de las lesiones retinianas de la enfermedad de Behçet.2
La prueba de patergia, una manifestación característica que presentan pacientes de la zona este del mediterráneo (positiva <30% en España) es la reactividad cutánea inflamatoria inespecífica, (infiltración por neutrófilos en ausencia de infección, seguida de la acumulación de células mononucleares) con formación de pápula o pústula eritematosa de más de 2mm de diamétro a las 24 a 48horas, tras el pinchazo con aguja estéril o suero salino <1cm de profundidad y con giros de la aguja en el antebrazo. Esta prueba puede ser también positiva en otras enfermedades, tales como el Síndrome de Sweet y el pioderma gangrenoso.2
El compromiso del sistema musculoesquelético existe en el 44–59% y se caracteriza por artralgias, mialgias y artritis asimétrica, mono–oliogoartritis, con predominio de grandes articulaciones de las extremidades inferiores (rodillas y tobillos) de evolución subaguda, intermitente y no deformante; se considera secundaria a sinovitis y trombosis de pequeños vasos. El inicio suele ser monoarticular y las reactivaciones oligoarticulares; de forma poco frecuente se presentan erosiones que a menudo son reversibles y pueden encontrarse datos de entesitis en las radiografías. Histológicamente hay infiltración de neutrófilos y células mononucleares en la sinovial.2
La afectación vascular tiene clara preferencia masculina. La enfermedad de Behçet, junto al lupus eritematoso sistémico y la enfermedad de Buerger, es una de las pocas vasculitis que pueden afectar tanto al árbol venoso como al arterial.6 Las características principales de esta afectación vascular son el implicar vasos de todos los tipos y tamaños, con acentuación del árbol venoso, los vasa nervorum están generalmente indemnes, los ANCA y otros anticuerpos están ausentes, lo mismo que los inmunocomplejos. A pesar de la tendencia a producir trombosis venosa el tromboembolismo es raro en esta enfermedad. La tromboflebitis superficial es la manifestación mas frecuente, a menudo asociada a punciones venosas; se presenta en 25% de pacientes. La trombosis venosa profunda ocupa el segundo lugar en frecuencia y afecta 10% de pacientes en las extremidades inferiores y puede provocar dermatitis crónica por estasis y ulceras. Otras complicaciones menos frecuentes son el síndrome de Budd–Chiari por trombosis de venas hepáticas o de la afectación cava. Las venas intracraneales también se afectan, reportándose trombosis venosa cerebral. La afectación arterial se presenta en 4%, y puede lesionar todas las arterias, pero tardíamente, tres a ocho años después del inicio de la enfermedad. El hallazgo histológico predominante es la vasculitis de los vasa vasorum. Los aneurismas pueden ser verdaderos o seudoaneurismas y tienen peor pronóstico que las trombosis. Pueden aparecer tras punciones arteriales y la cirugía es difícil, ya que suele reaparecer el aneurisma y ocasionar rechazos de la prótesis por fenómenos de patergia en la pared vascular. La enfermedad de Behçet es la única vasculitis que produce aneurismas pulmonares que se manifiestan por hemoptisis, son múltiples, bilaterales, se ven como opacidades en la radiografía, y su diagnóstico definitivo es por arteriografía, TAC o resonancia magnética.9
La afectación cardiaca con manifestaciones clínicas es rara, aunque de forma esporádica se han observado pericarditis, miocarditis, endocarditis, vasculitis coronaria, infarto de miocardio y aneurismas ventriculares, incluso lesiones inusuales como trombos intracardiacos o fibrosis endomiocárdica.
Las alteraciones del sistema nervioso central son, afortunadamente, poco frecuentes (5 a 15%) y ocurren en pacientes de mayor edad. Los signos neurológicos mas frecuentes son los síndromes de tronco cerebral y piramidales, la hipertensión intracraneal, la meningitis aséptica y los síndromes medulares. También se han descrito cuadros similares a esclerosis múltiple,7 La cefalea puede ser un dato de trombosis del seno dural. Los síntomas tienen exacerbaciones y remisiones y causan discapacidad progresiva; en etapa terminal la demencia se presenta en 30% de los pacientes. La meningitis aséptica aguda se desarrolla en las fases tempranas de la enfermedad, generalmente responde a esteroides y tiene buen pronóstico.2 Los datos recogidos del LCR son inespecíficos, aunque las concentraciones de proteína oligoclonales IgA e IgM pueden incrementar en fases activas y haber pleocitosis, con linfocitos. La RMN es útil en fases activas para los pacientes con afectación neurológica, pudiendo presentar lesiones focales múltiples hiperintensas en T2 en el cerebro, ganglios basales, y sustancia blanca, al igual que los pacientes con lupus eritematoso sistémico u otras conectivopatías.8

El involucramiento renal se informa en 3.8% de pacientes, la hematuria o proteinuria son los signos predominantes (10.8%) se ha identificado glomerulonefritis, las complicaciones mas frecuentes comunicados son enfermedad renal vascular por la formación de aneurismas de la arteria renal, estenosis o trombosis de la vena renal.1
La enfermedad de Behçet ha sido informada en asociación con síndrome mielodisplásico, hepatitis C, miositis aguda necrotizante, síndrome de Sweet y síndrome antifosfolipido. La presencia de anemia, trombocitopenia y células inmaduras en sangre periférica han sido identificadas en pacientes con enfermedad de Behçet. El examen de la médula ósea muestra mielodisplasia.2 La asociación de enfermedad de Behçet y mielodisplasia se ha identificado en un grupo de pacientes con una alteración citogenética común (trisomía 8) solo se han informado 10 casos en la literatura médica a nivel mundial y es más frecuente en población japonesa.3 El mecanismo se desconoce, pero la excesiva generación de especies reactivas de oxígeno por actividad de los neutrófilos, ha sido considerada como causa de daño tisular en pacientes con enfermedad de Behçet.
El síndrome mielodisplásico es caracterizado por disminución de la producción de especies reactivas de oxigeno por los neutrófilos, pero en los pacientes que tienen enfermedad de Behçet en fase activa se ha observado que los neutrófilos tienen un incremento de las especies reactivas de oxigeno, medidos por quimioluminicencia.11 Si la anemia con reticulocitos bajos y células dispoyéticas es sostenida en la enfermedad de Behçet, los médicos deberían estar alerta ante la posibilidad de síndrome mielodisplásico con aberración en el cromosoma 8, la cual se presenta en 10 a 20% en los pacientes con síndrome mielodisplásico no tratado y en 10 a 30% de pacientes con leucemia mieloide aguda de novo con aberraciones cromósomicas. Esto sugiriere que la trisomía 8 puede predisponer a enfermedad de Behçet a un subgrupo de pacientes con síndrome mielodisplásico, ya que el porcentaje de trisomía 8 en pacientes con síndrome mielodisplásico y enfermedad de Behçet es mayor que en aquellos que tienen síndrome mielodisplásico solamente. Las citopenias que se presentan son multifactoriales: fenómenos de autoinmunidad, hiperesplenismo, o empleo de quimioterapia.3 Otra causa que puede predisponer al desarrollo de síndrome mielodisplásico en Behçet es el uso de agentes inmunosupresores (agentes alquilantes) para tratar esta enfermedad, el riesgo de desarrollar mielodisplasia está relacionado a la dosis acumulada y a la duración del tratamiento con los diferentes agentes citotóxicos y la más frecuente asociación cromosómica involucra a los cromosomas 7 y 8. El citotóxico mas frecuentemente asociado es la ciclofosfamida, con dosis acumuladas >100g, pudiendo desarrollarse incluso 4 años después de suspender la terapia.12 El síndrome mielodisplásico secundario que presentó nuestro paciente fue anemia sideroblástica refractaria al tratamiento.
La hipercoagulabilidad es otra alteración hematológica que se identifica en un tercio de los pacientes con enfermedad de Behçet. Existe aumento en la generación de trombina, trombomodulina y alteración de la fibrinolisis. Otro factor involucrado es la hiperhomocistinemia. La tromboflebitis y la trombosis venosa profunda se identifican en 24% de los casos, aproximadamente. También puede ocurrir oclusión de grandes vasos y formación de aneurismas.1
Referencias
1. Yurdarkul S, Hamuryudan V, Yazici H. Behçet syndrome. Curr Opin Rheumatol 2003; 16:38–42. [ Links ]
2. Sakane T, Takeno M, Suzuki N, et al. Behçet disease. N Engl J Med 1999; 314:1284–1291. [ Links ]
3. Oh E, Yoon JS, Park YJ, et. al. Behçet Disease Associated with Myelodisplastic Syndrome: A Case Report. J Korean Med Sci 1999; 14:685–687. [ Links ]
4. Karuvannur S, Lipstein E, Brennessel D, et al. Atypical Behçet Syndrome in a patient with Myelodysplastic Syndrome. The Mount Sinai Journal of Medicine 2001; 68:403–405. [ Links ]
5. Yudarkul S. Tuzuner N, Yudarkul I, et. al. Gastrointestinal involvement in Behçet syndrome: a controlled study. Ann Rheum Dis 1996; 55:208–210. [ Links ]
6. Lie JT. Vascular involvement in Behçet disease: arterial y venous y vessels of all sizes. J Rheumatol 1992; 19:341 –343. [ Links ]
7. Serdaroglu P. Behçet disease and the nervous system. J Neurolol. 1998; 245:197–205. [ Links ]
8. Akman–Demir G, Kurt BB, Serdaroglu P, et. al. Seven years follow up of neurological involvement in Behçet syndrome. Arch Neurolol 1996; 53:691–694. [ Links ]
9. Hamuryudan V, Yurdakul S, Moral F, et. al. Pulmonary arterial aneurysms in Behçet syndrome: a report of 24 cases. Br. J Rheumatol 1994; 33:4–51. [ Links ]
10. Jorizzo JL, Abernethy JL, White WL, et. al. Mucocutaneus criteria for the diagnosis of Behçet disease: an analysis of clinicopathologic data from multiple international centers. J. Am Acad Dermatol 1995; 32:968–976. [ Links ]
11. Shyamala K, Lipsten E, Brennessel D, et. al. Atypical Behçet's Syndrome in a patient with Myelodisplatic Syndrome. The Mount Sinai Journal of Medicine 2001; 68:403–405. [ Links ]
12. Sirianni MC, Barbone B, Monarca B, et, al. Haematologica 2001; 86:1004–1005. [ Links ]
13. Chen KR, Kawahara Y, Miyakawa S, et. al. Cutaneous vasculitis in Behçet's disease: a clinical and histopathologic study of 20 patients. J Am Acad Dermatol 1997; 36:689–696. [ Links ]
14. Kim B, LeBoit PE. Histopathologic features of erythema nodosum–like lesions in Behçet disease: a comparison with erythema nodosum focusing on the role of vasculitis. Am J Dermatopathol 2000; 22:379–390 [ Links ]
15. Chun SI, Su WP, Lee S. et. al. Erythema nodusum–like lesions in Behçet's syndrome: a histopathologic study of 30 cases. J Cutan Pathol. 1989; 16:259–265. [ Links ]

