










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta médica de México
versión On-line ISSN 2696-1288versión impresa ISSN 0016-3813
Gac. Méd. Méx vol.142 no.1 Ciudad de México ene./feb. 2006
Artículo original
Las epidermolisis bullosas distróficas en México: 2470insG representa la mutación más común en 21 familias
2470insG, represents the commonest mutation in mexican patients with dystrophic bullous epidermolysis. A study of 21 families
Julio César Salas–Alanisa* y John A. McGrathb
a Servicios Médicos de la Universidad Autónoma de Nuevo León, Monterrey, NL, México
b Department of Cell and Molecular Pathology, St John's Institute of Dermatology, The Guy's King's College and St. Thomas' Hospitals Medical School, London, UK
Recibido en su versión modificada: 20 de enero del 2005
Aceptado: 09 de septiembre del 2005
*Correspondencia y solicitud de sobretiros:
Julio César Salas Alanis.
Otomie 206, Col. Azteca, Guadalupe, N.L.
CP 67150. México
Tel. (81) 8398–4080.
Correo electrónico: Correo electrónico.doctor@iuliosalas.com
Resumen
Antecedentes: Las epidermolisis ampollosas congénitas son enfermedades caracterizadas por ampollas en piel y mucosas al mínimo traumatismo. Son tres tipos: simple, unión y distrófica.
Las epidermolisis ampollosas distróficas (EAD) son causadas por mutaciones en el gen COL 7Al que codifica la producción del colágeno tipo VII localizado en las fibrillas de anclaje de la unión dermo–epidérmica.
Objetivo: Determinar las bases moleculares de las EAD en México.
Material y métodos: se analizaron ADN de 21 familias mexicanas con EAD. Se realizó reacción en cadena de la polimerasa, estudios de heteroduplex secuenciación de nucleótidos en ADN de los pacientes.
Resultados: Se detectó 59 de 67 mutaciones en 36 pacientes. Se encontraron seis mutaciones de tipo codón de terminación prematuro, substitución de glicina, remoción de intrones de novo y depleción interna.
La mutación comúnmente más encontrada fue la 2470insG, en 21 (58.35%) de 36 pacientes.
Conclusiones: En pacientes con EAD, la mutación 2470insG es la más frecuente en México. Recomendamos analizar esta mutación a Mexicanos con EAD como primera opción. Estos resultados son útiles para clasificar los subtipos de EAD, dar asesoramiento genético, así como para entender un poco más la fisiopatología de esta enfermedad mecano ampollosa.
Palabras clave: Epidermolisis bulosa distrófica, mutación 2470insG
Summary
Background: Type VII collagen gene (COL 7 Al) mutations are the cause of dystrophic epidermolysis bullosa (DEB), but most mutations are specific to individual families and there is limited data on the nature of COL 7 Al mutations in certain ethnic populations. Objective. To determine the molecular basis of DEB in Mexican patients and describe the most frequent mutation among this ethnic population.
Methods: Most subjects were approached at FUNDACION DEBRA MEXICO AC. Molecular analysis was performed by polymerase chain reaction (PCR) of genomic DNA using COL 7 A l–specific primers, heteroduplex analysis, and direct nucleotide sequencing.
Results: Fifty nine of 67 COL 7 Al possible mutations (88%) were identified; 36 individuals (31 recessive, five dominant) from 21 families. Recessive mutations included six frameshift mutations, four silent glycine substitutions and two splice site mutations.
Conclusions: The present study informs a different kind of mutation observed in our patient population. Only two mutations informed in this study had been described earlier among another ethnic group. The most frequent mutation was 2470insG, affecting 21(58.3%) out of 36 patients with DEB. These new data will be helpful in facilitating the accurate diagnosis of an DEB subtype, and will add further insight into the pathophysiology of this mechanobullous disease.
Key words: Dystrophic epidermolysis bullosa, 2470insG mutation
Introducción
Las enfermedades ampollosas que afectan la piel pueden ser genéticas o adquiridas. A nivel histológico, éstas pueden localizarse en la epidermis y/o membrana basal dermo–epidérmica. La membrana basal epidérmica representa una compleja red de moléculas de adhesión finamente relacionadas entre sí y con funciones específicas; cuando alguna de ellas falla, la fuerza de unión se pierde y provoca, en general, ampollas. Las enfermedades ampollosas adquiridas son debidas principalmente a agentes físicos (calor, fuego, radiación solar), químicos (agentes cáusticos) y menos frecuentes debido a anticuerpos circulantes IgGs, IgAs, IgMs y C3 (pénfigo y penfigoides) y/o infecciones (virus del herpes simple, Staphylococcus aureus).1
Las enfermedades genéticas que producen ampollas son variadas, sin embargo todas ellas afectan a una pequeña parte de la población. En Estados Unidos las estadísticas varían de acuerdo a cada enfermedad; en la EAD recesiva se estima un caso por cada 200,000 personas. Sin embargo, en México desconocemos por completo el porcentaje de niños que nacen con enfermedades genéticas ampollosas.2
Las epidermolisis ampollosas congénitas pueden heredarse con uno y otro patrones genéticos: recesivo y dominante, y probablemente representan las enfermedades ampollosas genéticas más comunes en México. Todas ellas presentan, en diferente grado de afección, ampollas y úlceras en piel y mucosas al mínimo traumatismo, de ahí su nombre de enfermedades mecano bulosas. Estas han sido divididas en forma general en tres grupos: simples, de unión y distróficas. En las formas simples la separación se forma por fractura debido a mutaciones en los filamentos de citoqueratinas 5 y 14 localizadas en la porción inferior de los queratinocitos basales de la epidermis. En las formas de unión y debido a mutaciones en la lámina 5, la ampolla se localiza en la lámina lúcida de la membrana basal epidérmica. Finalmente, las formas distróficas son por mutaciones en el colágeno VII, que forma las fibrillas de anclaje de la sub–lámina densa (Figura 1). Es imposible diferenciar al nacimiento el subtipo de epidermolisis, pues las tres formas muestran las mismas características: áreas de piel denudadas, erosiones y ampollas en la piel y mucosas.
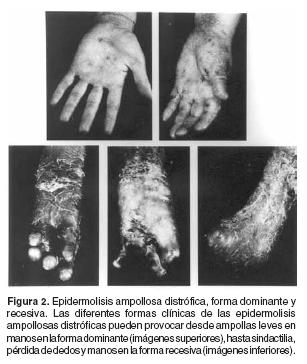
Las epidermolisis ampollosas distróficas se manifiestan por diferentes grados de afección, por ejemplo, las formas dominantes muestran pocas lesiones ampollosas y ulceraciones en la piel de las extremidades, principalmente en rodillas, pies, codos y manos; sin embargo, en las formas recesivas las lesiones pueden afectar grandes áreas del cuerpo y, con el tiempo, hasta provocar mutilaciones de dedos por cicatrización y absorción e incapacidades muy importantes (Figura 2). En estos pacientes, hay lesiones en las mucosas, provocando estenosis en el esófago, fisuras y hemorragia en el tracto intestinal (recto), ya que en todas las uniones epiteliales muestran pérdida de la adhesión. En algunos pacientes con úlceras crónicas es factible observar carcinomas epiteliales espinocelulares que, en estos pacientes, pueden provocar metástasis a edades tempranas.3
En la microscopía electrónica las fibrillas de anclaje formadas por colágenos VII y localizadas en la sub–lámina densa de la membrana basal epidérmica, presentan alteración en su estructura, disminución o inclusive ausencia total, sobre todo en las formas recesivas severas.4,5
El gen del colágeno VII (COL 7 Al) parece ser el gen con más exones conocido a la fecha, 118 exones y la detección de mutaciones es sin duda una tarea consumidora de tiempo, intensiva y costosa. La primera mutación informada en el gen del colágeno VII (COL 7 Al) fue identificada en pacientes con epidermolisis ampollosa distrófica recesiva en 1993.6 Desde entonces un sin número de mutaciones han sido publicadas alrededor del mundo y cada grupo étnico muestra en general mutaciones únicas; sin embargo, en ocasiones es factible observar mutaciones recurrentes.7–9
Algunas correlaciones geno/fenotipo han sido establecidas. Así por ejemplo, las formas recesivas mutilantes llamadas de Hallopeau–Siemens con ausencia total de colágeno VII en las fibrillas de anclaje, son generadas por mutaciones de codón de terminación prematura en ambos alelos. En las formas autosómicas dominantes de las epidermolisis ampollosas distróficas, la mutación consiste en una substitución de glicina dentro del triple hélix del colágeno VII provocando dificultad en el ensamblaje de las fibrillas y disminución de la fuerza de la unión dermo–epidérmica. Asimismo, en las formas menos severas, existe gran diversidad de mutaciones incluyendo mutaciones por pérdida, por remoción de intrón y mutaciones de codón de terminación prematura cerca del final 3', entre otras (Figura 3). El objetivo de este estudio fue analizar las mutaciones e informar la más frecuente en 21 familias mexicanas con epidermolisis ampollosas distróficas.
Material y métodos
Treinta y seis pacientes de 21 familias mexicanas con EAD fueron incluidos en este estudio. La mayoría de las muestras de los pacientes se obtuvieron a través de la Fundación DEBRA México A.C.10 Después de firmar la hoja de consentimiento informado, se obtuvo biopsia de piel para tinción con la técnica de Hematoxilina y Eosina; en algunos casos se realizó biopsia para estudios de microscopía electrónica y a todos los padres y enfermos se les extrajeron 6 mililitros de sangre. La mayoría de las familias analizadas provenían del noreste de México.
Prueba de la reacción en cadena de la polimerasa (PCR) y análisis Heteroduplex
Se extrajo ADN genómico de los linfocitos de sangre periférica según métodos convencionales, para obtener las secuencias del gen COL 7 A1, previa amplificación por PCR de los 118 exones (GenBank L23982, L02870). Asimismo, para generar estos productos de PCR, se utilizaron pares de cebadores de oligonucleótidos de los 118 exones del gen COL 7 A1.11,12
Para la técnica de PCR, se colocaron dentro de una centrífuga térmica Omni/Gene (Hybaid) 250 ng de ADN genómico, 6.25 pmol de cebadores, 37.5 nmol de MgCl2, 5 mmol de cada nucleótido y 1.25 U de polimerasa Taq (Perkin–Elmer) en 25 uL de volumen total. El rastreo de mutaciones fue realizado por análisis heteroduplex usando electroforesis en gel.13
Después del análisis de PCR, 3–8 uL de los productos de PCR fueron calentados a 98° C por 8 minutos, seguidos por 40 minutos a 68° C hasta conseguir la formación de los heteroduplex. Además, para detectar mutaciones homocigotas, volúmenes iguales de productos de PCR de individuos enfermos fueron mezclados con productos de PCR de controles normales antes de provocar la desnaturalización de ADN y posteriormente se realizó la electroforesis en gel. Los productos de PCR que mostraron bandas heteroduplex fueron purificadas usando columnas de rotación Qiaquik y secuenciados (en orientaciones 5'–3' y 3'–5') utilizando la secuenciación automática fluorescente ABI 310 (PE Biosystems).
Las mutaciones fueron verificadas por digestión con endonucleasas de restricción y/o por secuenciación directa automática del ADN amplificado de miembros de la familia y de individuos controles.
Resultados
La distribución de las 21 familias analizadas de los treinta y seis pacientes con epidermolisis ampollosas distróficas fue la siguiente: once de 21 (52.3%) familias eran de Nuevo León, ubicándose primordialmente en Villa de Santiago, Cadereyta, Montemorelos, Ciénega de Flores y Monterrey, todas ellas con patrón autosómico recesivo; dos familias de Zacatecas, dos de Matamoros, Tamaulipas; una familia de Veracruz, dos familias de Monclova, Coahuila y finalmente una familia de Guadalajara, Jalisco. De los 36 pacientes con EAD, 31 tenían patrón recesivo y sólo cinco tenían patrón dominante. En una familia existieron uno y otro patrones de herencia afectando a un miembro con patrón recesivo y siendo el padre y una hermana con patrón dominante.
Se detectaron 59 (88%) de 67 posibles mutaciones en el gen COL 7 A 1. De los 31 pacientes con epidermolisis ampollosa distrófica recesiva, 16 (51 %) fueron homocigotos para el codón de terminación prematura (mutación frameshift) en el exón 19 del gen 9 COL 7 Al, llamada 2470insG/2470insG (Figura 4). Cinco pacientes fueron heterocigotos en combinación con diferentes mutaciones en otros alelos. Entre estos 21 pacientes de las 12 familias que comparten la misma mutación, sólo 2 familias se conocían como familiares, sin embargo, la mayoría de ellos convivían en una zona rural localizada al sur del estado de Nuevo León. Los estudios en haplotipos sugieren una propagación común ancestral.14 Esta mutación constituye la primera mutación recurrente en México y representa la mutación más frecuente en el mundo en pacientes con patrón recesivo de la epidermolisis ampollosa distrófica. Así, por ejemplo, R578X, 7786delG y R2812X, parecen ser exclusivas de la población británica, mientras que la mutación 5818delC, 6573=IG–C y E2857X corresponden a la población japonesa.15

La mutación en la lectura del marco genético provoca un codón de terminación prematuro en el exón 19 con la sucesiva ausencia de producción del colágeno VII para formar las fibrillas de anclaje en la sublámina densa de la unión dermo–epidérmica, provocando clínicamente en los pacientes, grandes ampollas y úlceras al mínimo traumatismo en la piel y membranas mucosas, mutilaciones semejantes al cuadro de Hallopeau Siemens. Sin embargo, estos pacientes tienen sólo fragilidad cutánea leve y carecen de pseudodactilia, cicatrizaciones anormales, incluso hay pacientes homocigotos mayores de 70 años sin cáncer cutáneo.
La segunda mutación más común correspondió a 6863 del 16 afectando a varias generaciones de pacientes de Monclova, Coahuila con patrón dominante. Cabe mencionar que en forma general, las formas autosómicas dominantes se deben a sustituciones de glicina en la triple hélice, sin embargo en estas familias, la mutación se debió a una deleción de 16 pares de bases de nucleótidos, provocando una pérdida de la lectura del marco genético por generación de codón de terminación prematura, resultando en la exclusión del exón 87 con un fenotipo dominante. Las manifestaciones clínicas de estos pacientes incluían pocas lesiones ampollosas en sitios de roce, nulo o muy poca afección de mucosas y ausencia de cicatrices deformantes o sindactilias. En esta mutación, la pérdida de 16 pares de bases provocó la producción menor de colágeno VII. Dicha mutación se localiza en el exón 86.16
Otras mutaciones encontradas en este estudio son cinco pequeñas inserciones o deleciones en COL 7 Al. Estas son; 3948insT, 4429de1G, 5572deIG, 5048insGAAA y 6691 insC. De éstas, sólo la primera fue informada previamente por nuestro grupo en 1998.17 Estas mutaciones provocan que la producción de colágeno VII sea de mala calidad o muy poca, debido a un codón de terminación prematura en la lectura del marco genético. Clínicamente, estos pacientes presentaron signos clínicos semejantes, ampollas generalizadas sin cambios mutilantes importantes y sin daño a las membranas mucosas. Cabe mencionar que tres de estas mutaciones fueron heterocigotas, combinadas con sustituciones silenciosas de glicina en el otro alelo del gen COL 7 A1 (4429deIG/G 1782R; 5772de1G/G 1703E; 6691 insC/G 169E). Todas estas mutaciones no habían sido descritas en otros grupos étnicos a la fecha. Además de las mutaciones informadas anteriormente, encontramos una paciente con distrofia ungueal así como moderadas cicatrices en la espalda orientando el diagnóstico a epidermolisis ampollosa distrófica dominante. Dicha paciente tiene padres y 12 hermanos clínicamente y genéticamente sanos, en ella detectamos la mutación por substitución de 11–glicina, G2043R. Esta mutación está informada como la causa más frecuente de epidermolisis ampollosa distrófica dominante en todo el mundo, sin embargo, en esta paciente, no se encontó la mutación en ningún miembro de la familia, por lo tanto dicha mutación se considerada de novo.18
Mutación de patrón recesivo y dominante en una misma familia. Uno de los casos más fascinantes en los resultados encontrados fue sin duda una familia con antecedentes de epidermolisis ampollosa distrófica dominante en la cual el padre y una hija mostraron distrófica ungueal, cicatrices irregulares en rodillas, codos y manos, además de ausencia de lesiones mutilantes, sindactilia o lesiones en las mucosas. En esta familia, el último hijo en nacer presentó signos típicos de una epidermolisis ampollosa distrófica con patrón recesivo tipo Hallopau Siemmens, mostrando gran cantidad de ampollas en forma generalizada, con afección a mucosas, sindactilia y mutilación de algunos dedos (Figura 5). El análisis genético de esta familia mostró una mutación por deleción interna (6863del16) en padre e hija enferma y en el varón con signos clínicos característicos de epidermolisis ampollosa distrófica recesiva severa una mutación por empalme 425 A–to–G heredada de la madre.16

Discusión
Los beneficios inmediatos de conocer las diferentes mutaciones que afectan a los pacientes mexicanos con epidermolisis ampollosa distrófica consisten en otorgar información del grado de patrón genético de cada familia para posteriores embarazos y además, la posibilidad de realizar biopsia de vellosidades coriónicas en las primeras semanas del embarazo para el diagnóstico temprano de la enfermedad. Es un hecho que en algunos países del mundo se está realizando el diagnóstico en DNA pre–implantación en este tipo de enfermedad.19
Los hallazgos informan que la mutación más común en México es sin duda la homocigoto 2470insG/2470insG, afectando a 15 de los 36 pacientes analizados (41.6%), provocando una forma moderada de epidermolisis ampollosa distrófica recesiva a pesar de corresponder a una mutación de codón de terminación prematura en ambos alelos. El primer paciente con esta mutación fue reportado en un mexicano radicado en Los Ángeles, California en los Estados Unidos, mostrando una combinación heterocigota 2470insG/385deIG. A la fecha y con los hallazgos de esta investigación, ya son 22 pacientes en total que muestran en forma heterocigota u homocigota esta mutación. Esta información es importante para iniciar la detección de mutaciones en pacientes del norte de México que padezcan epidermolisis ampollosa distrófica recesiva. Asimismo, cabe mencionar que los individuos con esta mutación homocigota padecen la enfermedad de afección moderada, cuando se les compara con aquellos que muestran una mutación heterocigoto. Esta alteración molecular ya ha sido publicada previamente.20
Cabe mencionar que algunas pacientes mujeres con esta mutación homocigota han procreado hijos con partos vaginales sin ninguna complicación posterior de índole tal como cicatrices, bridas o infecciones severas. Además, a la fecha ningún paciente con este tipo de mutación no ha desarrollado cáncer de piel ni siquiera en edades adultas, el cual es común desarrollar en este tipo de pacientes.
Un beneficio de estos estudios radica además en determinar que individuos de la misma comunidad son portadores de esta mutación, debido al riesgo y posibilidad de matrimonios consanguíneos. Estableciendo grupos como DEBRA México A.C., no sólo se facilita el trabajo en la obtención de muestras para detectar mutaciones, sino que además, apoya a los padres y al paciente en la comprensión y manejo de la enfermedad.
Referencias
1. Arenas R. Dermatología, Atlas, diagnóstico y tratamiento, segunda edición Interamericana McGraw–Hill 1999; 123–150. [ Links ]
2. Fine JD, Bauer EA, Briggaman RA et al. Epidermolysis Bullosa. Application of epidemiologic principles to the study of a group of rare disease via a disease registry. Dermatol Clinics 1995; 13:659–670. Revised clinical and laboratory criteria for subtypes of epidermolysis bullosa. A consensus report by the subcommittee on Diagnosis and Classification of the National Epidermolysis Bullosa Registry. J Am Acad Dermatol 1991; 24:119–135. [ Links ]
3. Fine JD, Eady RAJ, Bauer EA, Briggaman RA, Bruckmer–Tuderman L, Christiano AM, et al. Revised classification system for inherited epidermolysis bullosa: Report of the second International consensus meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol 2000;42:1051–66. [ Links ]
4. Giles SM, Mcgrath JA, Richards AJ, Christiano AM, Uitto J, Michael Pope F, Lady JA. Clinicopathological Correlations of Compound Heterozygous COL 7 Al Mutations in Recessive Dystrophic Epidermolysis Bullosa. J Invest Dermatol 1996; 107: 171–177. [ Links ]
5. Heagerty AHM, Kennedy AR, Leigh M, Purkis P, Lady RAJ. Identification of epidermal basement membrane defect in recessive dystrophic epidermolyisis bullosa by LH7–2 monoclonal antibody: use in diagnosis. Br J Dermatol 1986; 115:125–131. [ Links ]
6. Christiano AM, Greenspan DS, Hoffman GC et al. A non sense mutation in type VII collagen in two affected siblings with recessive dystrophic epidermolysis bullosa. Nat Genet 1993; 4:62–66. [ Links ]
7. Mellerio JE, Giles SM, Allison W, Ashton GH, Christiano AM, Uitto J, Eady RA, McGrath JA. Recurrent mutations in the Type VII Collagen Gene (COL 7 Al) in patients with Recessive Dystrophic Epidermolysis Bullosa. J Invest Dermatol 1997; 109:246–249. [ Links ]
8. Shimizu H, McGrath JA, Christiano AM et al. Molecular basis of recessive dystrophic epidermolysis bullosa; genotype/phenotype correlation in a case of moderate clinical severity. J Invest Dermatol 1996; 106:119–124. [ Links ]
9. Hovnanian A, Rochet A, Bodemer C. et al. Characterization of 18 new mutations in COL 7 A1 in recessive dystrophic epidermolyisis bullosa provides evidence for distinct molecular mechanisms underlying defective anchoring fibril formation. Am J Human Genet 1997; 61:599–610. [ Links ]
10. Salas–Alanis JC. Nace Fundación DEBRA MEXICO AC, Carta al editor, Rev Mex Dermatol 1998; 42:172–193. [ Links ]
11. Christiano AM, Hoffman G, Chung–Honet LC et al. Structural organization of the human type VII collagen gene (COL 7 Al) compromised of more exon than any previously characterized gene. Genomics 1994;21:169–179. [ Links ]
12. Christiano AM, Hoffman GC, Zhang X. et al. A strategy for identification of sequence variants in COL 7 Al, and a novel 2 bp deletion mutation in recessive Dystrophic epidermolysis bullosa, Hum Mutant 1997; 10:408–414. [ Links ]
13. Ganguly A, Rock MJ, Prockop DJ. Conformation–sensitive gel electrophoresis for rapid detection of single–base differences in double–stranded PCR products and DNA fragments; evidence for solvent–induced bends in DNA heteroduplex. Proc Natl Acad Sci USA 1993; 90:10,325–329. [ Links ]
14. Salas–Alanis JC, Mellerio JE, Amaya–Guerra Mario et al,. Frameshift mutations in the type VII collagen gen (COL 7 Al) in five Mexican cousins with recessive Dystrophic epidermolysis bullosa. Br J Dermatol 1998; 138:852–858. [ Links ]
15. Murata T, Masunaga T, Shimizu H, Nishikawa T. Differences in recurrent COL 7 Al mutations in dystrophic epidermolysis bullosa: ethnic–specific and worldwide recurrent mutations. Arch Dermatol Res 2004. [ Links ]
16. Cserhalmi–Friedman MB, Mcgrath JA, Mellerio JE, Romero R, Salas–Alanis JC, et al. Restoration of an Open Reading Frame Resulting from Skipping orfan Exon with an Internal Deletion in the COL 7 Al Gene. Lab Invest 1998; 78:1483–1492. [ Links ]
17. Salas–Alanis JC, Mellerio JE, Amaya–Guerra M, et al. Frameshift mutation in the type VII Collagen gene (COL 7 Al) in five Mexican cousins with recessive dystrophic epidermolysis bullosa. British J Dermatol 1998; 138:852–858. [ Links ]
18. Mellerio JE, Salas–Alanis JC, Talamantes ML, et al. A recurrent glycine substitution mutation, G2043R, in the type VII collagen gene (COLA71) in dominant dystrophic epidermolysis bullosa. Br J Dermatol 1998; 139:730–737. [ Links ]
19. McGrath JA, Dunnill MGS, Christiano AM, Lake BD, Atherton DJA, Rodeck CH, et al. First trimester DNA–based exclusion of recessive dystrophic epidermolysis bullosa from chorionic villus sampling. British J of Dermatol 1996; 134:734–739. [ Links ]
20. McGrath JA, Ashton GHS, Mellerio JE, et al. Moderation of phenotype severity in Dystrophic and functional forms of epidermolysis bullosa trough in–frameshift mutations. J Invest Dermatol 1999; 113:314–321. [ Links ]

