










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkGaceta médica de México
versão On-line ISSN 2696-1288versão impressa ISSN 0016-3813
Gac. Méd. Méx vol.141 no.6 Ciudad de México Nov./Dez. 2005
Ejercicio clínico patológico
Varón adolescente con dolor en el muslo derecho y dificultad para la deambulación
A sisteen year–old male with right calf pain and motor difficulty
Francisco Uc–Pool,ª Sergio Torres–Vargas,b e Isabel Alvarado–Cabrerob*
ª Hospital de Especialidades, y
b Hospital de Oncología, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, México, D. F., México
Coordinador: Dr. Manuel de la Llata Romero
Colaboradores: Dr. Juan Urrusti Sanz
Dr. Jesús Aguirre García
Dr. Roberto Sánchez Ramírez
*Correspondencia y solicitud de sobretiros:
Dra. Isabel Alvarado–Cabrero.
Servicio de Anatomía Patológica, Hospital de Oncología, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social.
Av. Cuauhtémoc 330, Col. Doctores, Delegación Cuauhtémoc,
06525, Teléfono: 5627 6900, ext. 21355.
Correo electrónico: Isa98@prodigy.net.mx
Presentación del caso
Se trató de un joven del sexo masculino de 16 años de edad, con antecedentes de carcinoma broncogénico y ce rvi co uteri no en su abuelo y abuela maternos respectivamente, tabaquismo y etilismo negativos y sin enfermedades previas de importancia. Inició su padecimiento en noviembre de 2004 con dolor en el muslo derecho, de tipo punzante, que se irradiaba a todo el miembro pélvico y dificultad para la deambulación, por lo que acudió a un facultativo.
En la exploración física se detectó aumento de volumen en la cara anterior del muslo derecho, indicándose estudios de imagen (placa simple de huesos y resonancia magnética). En la placa simple se observó el fémur íntegro y una lesión localizada en los tejidos blandos. La resonancia magnética mostró un tumor de 10 x 8 cm en el compartimiento anterior, tercio superior del muslo derecho, en estrecho contacto con el fémur y con los vasos femorales.
La telerradiografía de tórax y el ultrasonido hepático no mostraron actividad tumoral.

Diagnóstico
El diagnóstico inicial fue de neoplasia originada en el fémur, con dos posibilidades, sarcoma osteogénico o sarcoma de Ewing. Sin embargo, los estudios de imagen mostraron que el hueso estaba respetado y que la neoplasia tenía como asiento los tejidos blandos del muslo, por lo que la segunda consideración diagnóstica fue de rabdomiosarcoma. La biopsia de la lesión reveló una neoplasia compuesta por una población monótona de células pequeñas, redondas y basófilas, con crecimiento difuso, por lo que las opciones de diagnóstico fueron: sarcoma de Ewing extraesquelético, linfoma o rabdomiosarcoma. Debido a la similitud morfológica de estas neoplasias, fue necesario efectuar un panel de inmuno–histoquímica que incluyó los siguientes marcadores: CD99, CD45ro, sinaptofisina, vimentina, desmina, Myod1 y Tdt. La neoplasia fue positiva para CD99 y sinaptofisina, por lo que se concluyó que se trataba de un sarcoma de Ewing (SE) extraesquelético.
En el mes de enero del siguiente año se inició tratamiento con un esquema de quimioterapia a base de vincristina, etopósido y cisplatino, y al no haber respuesta tumoral, se cambió el esquema por ifosfamida y VP16. Con este último tratamiento se obtuvo una respuesta de reducción tumoral de 30%. Los estudios de extensión no mostraron enfermedad a distancia.
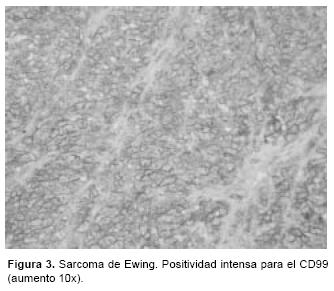
En marzo del 2005 se le practicó resección compartamental anterior amplia. En el Servicio de Anatomía Patológica se recibió un espécimen de 8 x 6 cm de bordes irregulares, formado en su porción externa por músculo. En los cortes seriados prácticamente todo el espécimen estaba constituido por una neoplasia blanquecina, de 4 x 5 cm, con aspecto de "carne de pescado", blanda, con focos de hemorragia, y con bordes infiltrantes. En el estudio microscópico se observó un aspecto morfológico diferente al de la biopsia inicial, conformado por células grandes, con citoplasma eosinófilo amplio y núcleos centrales pleomórficos, planteándose la posibilidad diagnóstica de rabdomiosarcoma (Figuras 1 y 2). Debido a la disparidad en las morfologías del tumor, se efectuó un nuevo panel de marcadores, resultando negativo a marcadores musculares como desmina, MyoD1 y mioglobina, por lo que se descartó rabdomiosarcoma. Por otro lado, el CD99, el WT–1 y la sinaptofisina, fueron positivos (Figura 3), por lo que se hizo el diagnóstico final de SE con cambios morfológicos secundarios al tratamiento por quimioterapia. El tratamiento posterior fue con radioterapia en el sitio de resección tumoral (Figura 4).



Comentario
El sarcoma de Ewing es una neoplasia de la infancia que ocupa el segundo lugar entre los tumores óseos de esa edad. El SE extraesquelético se origina en tejidos blandos, tronco o extremidades; la edad al momento del diagnóstico tiene un amplio margen, desde la infancia hasta la vejez. Dada la rareza de este padecimiento se cuenta con pocos estudios clínicos.1,2 Los sarcomas verdaderos de hueso y tejidos blandos, como el osteosarcoma de células pequeñas y el rabdomiosarcoma, pueden confundirse con el SE, lo que debe evitarse ya que no son incluidos en los protocolos de tratamiento del SE. Sin embargo, el error diagnóstico es común, ya que mediante microscopia de luz el diagnóstico diferencial de los tumores de células pequeñas y basófilas puede resultar imposible.1 El panel de inmunohistoquímica obligado en el diagnóstico de estas neoplasias debe incluir los siguientes anticuerpos: CD99 y sinaptofisina, que son positivos en el SE, MyoD1 y desmina, que son positivos en el rabdomiosarcoma, antígeno leucocitario común, positivo en los linfomas y TdT, positivo en la leucemia y linfoma linfoblástico.3–4 Algunos estudios indican que sólo la localización anatómica y la extensión de la lesión parecen influir en el comportamiento de la enfermedad, aunque en general, se acepta que ni las variables de factor pronóstico, ni el tratamiento óptimo han sido bien definidos.3–5
Con el advenimiento de la quimioterapia multiagente combinada, la cirugía y la radiación, el pronóstico del SE ha mejorado dramáticamente desde la década pasada. En los años 80 del siglo pasado, se observó que el tratamiento con ifosfamida solo, o en combinación con etopósido, es altamente efectivo en pacientes con sarcomas recurrentes.2,6–8 El caso presentado no respondió a los esquemas de quimioterapia utilizados; algunos tumores son refractarios a la quimioterapia preoperatoria o a la manipulación hormonal, lo que puede deberse a una gran variedad de factores, tales como resistencia intrínseca a los fármacos (por ejemplo, expresión del factor de resistencia a múltiples fármacos, desregulación de la vía de la apoptosis) o al acceso inadecuado de la quimioterapia al tumor.9,10
En la mayoría de los casos, la manipulación hormonal o con quimioterapia de los tumores provoca cambios morfológicos en los mismos, tales como degeneración o muerte celular. La extensión del daño es muy variable, debido a la gran heterogeneidad de las células tumorales en una misma neoplasia. Se pueden observar cambios morfológicos tales como necrosis coagulativa extensa —en fases iniciales con formación de "fantasmas celulares"—, así como fibrosis del estroma de sostén. Por otro lado, como sucedió en el caso que presentamos, las células pueden mostrar cambios pleomórficos con núcleos grandes, de apariencia "grotesca" e invaginaciones irregulares. Es importante conocer este fenómeno, ya que la morfología de una neoplasia puede ser completamente distinta a la original, sugiriendo un diagnóstico que podría ser erróneo.11
Conclusiones
Siempre que se estudie una neoplasia de células pequeñas, redondas y basófilas, es preciso utilizar un panel amplio de anticuerpos, lo que aunado al estudio clínico adecuado del paciente llevará a diagnósticos más precisos. El sarcoma de Ewing es una neoplasia agresiva que puede ser refractaria al tratamiento. La quimioterapia puede ocasionar importantes cambios morfológicos en las neoplasias, de manera que su morfología inicial puede transformarse considerablemente.
Referencias
1. Hartmann K, Triche T, Kinsella T, Miser J. Prognostic value of histopathology in Ewing's Sarcoma. Cancer 1991; 67:163–171. [ Links ]
2. Ahmad R, Mayol B, Davis M, Roggraff B. Extraskeletal Ewing's Sarcoma, Cancer 1999; 85:721–731 [ Links ]
3. Giu M, Antonescu C, Guiter G, Huvos A, Landayi M, Zakowski M. Cytokeratin immunoreactivity in Ewing's Sarcoma. Am J Surg Pathol , 2000; 24:410–416. [ Links ]
4. Contesso G, Llombart–Bosch A, Terrier P, Peydro–Olaya A, Henry–Amai M, Oberlin O, et al. Does malignant small round cell tumor of the thoracopul–monary region (Askin Tumor) constitute a clinicopathologic entity? Cancer 1992; 69:1012–1020. [ Links ]
5. Kumar S, Pack S, Kumar D, Walker R, Quezada M, Zhuang Z, et al. Detection of EWS–FLI–1 fusion in Ewing's Sarcoma/Peripheral Primitive Neuroectodermal Tumor by fluorescence in situ hybridization using formalin–fixed paraffin–embedded tissue. Human Pathol 1999; 30:324–330. [ Links ]
6. Collini P, Sampietro G, Poertulli R, Luksch R, Casali P, Mezzelani A, et al. Cytokeratin immunoreactivity in 41 Cases of ES/PNET confirmed by molecular diagnostic studies. Am J Surg Pathol 2001 ;25:273–274. [ Links ]
7. Pahrman D, Hijazi Y, Steinberg S, Meyer W, Horowitz M, Chin–Yuan T, et al. Neuroectodermal differentiation in Ewing's Sarcoma family of tumors does not predict tumor behavior. Human Pathol 1999; 30:911–918. [ Links ]
8. Wexler L, DeLaney T, Tsokos M, Ávila N, Steinberg S, Weaver–McClure L, et al. Ifosfamide and etoposide plus vincristine, doxorubicin, and cyclophosphamide for newly diagnosed Ewing's Sarcoma family of tumors. Cancer 1996; 78:901–911. [ Links ]
9. Riopel M, Dickman P, Link MS, Perlman E. MIC2 analysis in pediatric lymphomas and lekemias. Hum Pathol 1994; 25:396–399. [ Links ]
10. GUM, Antonescu CR, Guiter G. Cytokeratin immunoreactivity in Ewing's Sarcoma. Prevalence in 50 cases confirmed by molecular diagnostic studies. Am J Surg Pathol 2000; 24:410–416. [ Links ]
11. Delatre O, Zucman J, Melor T. The Ewing family of tumors–a subgroup of small round cell tumors defined by specific chimeric transcripts. N Engl J Med 1994; 331:294–299. [ Links ]

