










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta médica de México
versión On-line ISSN 2696-1288versión impresa ISSN 0016-3813
Gac. Méd. Méx vol.141 no.6 Ciudad de México nov./dic. 2005
Artículo original
Linfoma primario del sistema nervioso central en pacientes inmunocompetentes
Primary central nervous system lymphoma in immunocompetent patients
Gerardo Guinto–Balanzar,ª* Ignacio Félix–Espinoza,b Salvador De Anda Ponce–de –León,ª Norma C. Aréchiga–Ramos,c Víctor Arteaga–Larios,ª y Kalman Kovacsd
ª Servicios de Neurocirugía,
b Neuropatología, y
c Neurología, Hospital de Especialidades del Centro Médico Nacional Siglo XXI, México, D.F., México
d Servicio de Neuropatología, St. Michael's Hospital en Toronto, Canadá.
*Correspondencia y solicitud de sobretiros:
Dr. Gerardo Guinto Balanzar.
Servicio de Neurocirugía del Hospital de Especialidades del Centro Médico Nacional Siglo XXI.
Cuauhtémoc 330, Colonia Doctores,
México, D.F.
Teléfonos: 5627 6900 extensión 21492, 5568 6734 y 5568 6957. Fax 5135 1778.
Correo electrónico: gguinto@prodigy.net.mx
Recibido en su versión modificada: 24 de febrero de 2005
Aceptado: 1 de abril de 2005
Resumen
El linfoma primario del sistema nervioso central ha sido informado con frecuencia en pacientes que padecen síndromes de inmuno deficiencia. Sin embargo ésta no es una condición necesaria para su presentación, dado que existen informes de la enfermedad en sujetos inmunológicamente competentes.
En el presente trabajo se analizaron en forma retrospectiva, los expedientes de 22 pacientes inmunocompetentes con diagnóstico confirmado de linfoma primario encefálico, se revisó la literatura mundial, con el fin de analizar objetivamente las manifestaciones clínicas, comportamiento radiológico, aspecto histopatológico, dificul tades diagnósticas y terapéuticas, así como las consideraciones pro nósticas.
El promedio de edad fue de 65 años y con una relación equitativa hombre/mujer. El tiempo de evolución del cuadro clínico fue de 80.4 días y estuvo dominado por cefalea y déficit neurológico focal. En cuatro pacientes se encontraron lesiones múltiples, mientras que en el resto se trataba de lesiones únicas con localización predominante en la región periventricular de los hemisferios cerebrales. Todos los pacientes fueron manejados inicialmente con esteroides y sometidos a toma de biopsia por estereotaxia. La variedad histológica más frecuente fue la de células grandes difusas y la totalidad de los casos reaccionaron positivamente a antígenos de células B en la inmunohistoquímica. Los 22 pacientes fueron tratados con radio terapia y 10 de ellos además con quimioterapia con metotrexato. La supervivencia promedio fue de 11 meses en los pacientes radiados y de 36 meses en los que se agregó quimioterapia.
Palabras clave: Biopsia estereotáctica, linfoma primario del sistema nervioso central, quimioterapia, radioterapia, tumor cerebral.
Summary
Primary central nervous system lymphoma has been traditionally described in patients with immunodeficiency syndromes; however, there is an increasing number of immunocompetent patients with this type of tumor that have been reported recently. In this paper we have retrospectively analyzed 22 immunocompetent patients with a confirmed diagnosis of primary lymphoma of the brain. The mean age in this group was 65 years with a similar male/female ratio. The time of evolution of the clinical course was 80.4 days and it was mainly characterized by headache and focal neurological deficit. In four patients multiple lesions were observed, while the remaining presented single lesions mainly located in the periventricular area of the cerebral hemispheres. All patients were initially administered steroids and a stereotactic biopsy was performed. The majority of tumors were histologically classified as diffuse large cells and all of them showed a positive reaction to B cells antigens on immunohistochemistry. All patients were treated with radiotherapy and in 10 of them, chemotherapy with methotrexate was also indicated. The mean survival rate was 11 months among patients treated with radiotherapy alone and increased to 36 months when chemotherapy was added.
Key words: Brain tumor, chemotherapy, primary central nervous system lymphoma, radiotherapy, stereotactic biopsy.
Introducción
En 1929, Bailey1 describió un tumor histológicamente idéntico al linfoma primario del sistema nervioso central (LPSNC) al que denominó sarcoma perivascular; sin embargo, fue a partir de 1974 cuando esta neoplasia fue reconocida como una entidad patológica distinta.2 Se trata de una forma rara de linfoma extranodal confinada al encéfalo y que ha sido encontrado en especial en pacientes que padecen síndromes de inmunodeficiencia, ya sea congénitos o adquiridos. Sin embargo en fechas recientes, el número de casos que se presentan en pacientes inmunocompetentes (IC) va en aumento, quienes a pesar de tener un patrón histopatológico similar, muestran aspectos patogénicos y clínicos muy diferentes, en especial relacionados con factores pronósticos y terapéuticos.3
El sistema nervioso central es generalmente considerado como un santuario inmunológico, por lo que resulta difícil entender la causa por la que existan linfomas que primariamente aparecen en este sitio. Para explicar lo anterior se ha planteado la posibilidad de que en estos pacientes exista un determinado proceso (hasta el momento desconocido), que atraiga a las células linfáticas sanguíneas y las estimule a proliferar localmente y presentar una selección clonal.4 En otras palabras, parecería que en estos casos el cerebro pierde algún mecanismo regulador de la proliferación linfática que está normalmente presente en los tejidos periféricos.5
Se han postulado tres hipótesis para explicar esto:61) las células B se pueden transformar en alguna parte del organismo expresando moléculas de adhesión específicas para el endotelio cerebral; 2) las células del linfoma pueden ser sistemáticamente erradicadas por un sistema inmune intacto, pero relativamente protegidas dentro del sistema nervioso central; 3) una lesión inflamatoria intracerebral policlonal puede ser el primer paso, la cual se expande y progresa a un estado neoplásico monoclonal originando este tumor. El análisis citogenético de estos tumores ha demostrado anormalidades clonales en los cromosomas 1, 6, 7 y 14, así como traslocaciones (1;14), (6;14), (13;18) y (14;21).
El LPSNC representa 1 a 6% de todas las neoplasias intracraneales y 1 a 2% de los linfomas extranodales.7 Aun cuando la frecuencia de presentación del síndrome de inmunodeficiencia adquirida (SIDA) se ha incrementado notablemente en la actualidad, la incidencia global de este tumor ha disminuido.8 Por otro lado, con respecto al LPSNC que se presenta en pacientes IC, éste con mayor frecuencia se diagnostica en pacientes mayores de 60 años y presenta hallazgos clínicos y radiográficos que sugieren que se trata de una entidad patológica distinta.
Existen algunos factores de riesgo que se han relacionado con mayor frecuencia a la aparición del LPSNC en pacientes IC, los cuales incluyen: enfermedades autoinmunes, infecciones por virus de la hepatitis C, virus linfotrópico de células T humanas (HTLV) I/II o Helicobacter pylori.9 De igual manera, ha sido informado que 4.5 a 8% de los pacientes presentan historia genética de cáncer, en especial leucemia o adenocarcinoma y que este antecedente ha sido referido como un indicador de mal pronóstico. Finalmente, a diferencia del LPSNC relacionado con el SIDA, el que se presenta en los pacientes IC casi nunca se relaciona con infección del virus Epstein–Barr.10,11
En los estudios de imagen, el LPSNC se manifiesta por lesiones únicas o múltiples, generalmente en la región peri–ventricular y éstas muestran diferentes patrones de reforzamiento con el medio de contraste, dado que tiende a ser anular en casos de pacientes con inmunodeficiencia, mientras que es homogéneo en los IC. Sin embargo, aun cuando los estudios de imagen sean altamente sugestivos del diagnóstico, es necesaria la confirmación histológica. Debido al tamaño y la localización de la mayoría de las lesiones en la profundidad del parénquima cerebral, es innegable que el mejor método para obtener el diagnóstico es la biopsia por estereotaxia.12,13 Sin embargo, este procedimiento presenta algunos problemas: en primer lugar, en ocasiones no es posible obtener una cantidad suficiente de tejido para efectuar el diagnóstico correcto; en segundo lugar, la mayoría de los pacientes con estas lesiones ya han sido tratados con esteroides antes de la cirugía, medicamentos que han demostrado tener un efecto citotóxico en los linfomas cerebrales, condicionando una reducción en el tamaño o hasta desaparición del tumor. Este hecho dificulta no sólo el diagnóstico histológico sino la toma de biopsia en sí misma.
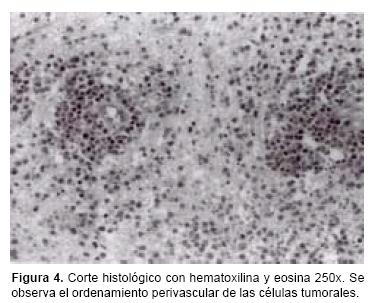
Los estudios histopatológicos han demostrado que los LPSNC en pacientes IC son muy similares a los que se presentan en los pacientes VIH–positivos. Con microscopia a menor aumento se demuestra el patrón de infiltración angiocéntrico, formándose collares de células tumorales alrededor de los pequeños vasos sanguíneos con diferentes cantidades de depósitos de reticulina.14 En casos avanzados se llega a perder este ordenamiento perivascular y las células tienden a invadir el parénquima cerebral circundante, ya sea en forma de compactos agregados o como una infiltración difusa de células tumorales similar a la encefalitis.15 Se ha informado con frecuencia reacción astrocítica que en ocasiones tiende a ser muy marcada,16 así como áreas de necrosis coagulativa. Todo esto explica el motivo por el cual con mucha frecuencia estas lesiones son confundidas en el estudio transoperatorio con astrocitomas, glioblastomas multiformes o metástasis.17
Con respecto al tratamiento del LPSNC en pacientes IC se ha demostrado que la resección quirúrgica de la lesión o lesiones no contribuye a la supervivencia;18,19 de hecho, la resección parcial representa un factor pronóstico desfavorable. Es por ello que la cirugía en estos pacientes se reduce a establecer el diagnóstico mediante una toma de biopsia, preferentemente por método estereotáctico, dado que se trata como ya se mencionó, de lesiones profundas.
Aunque el LPSNC es marcadamente radiosensible, cuando esta modalidad terapéutica se usa en forma aislada, ocurre que más de 80% de los pacientes presentan recurrencia dentro de los primeros 10 a 14 meses con un porcentaje de supervivencia a cinco años de 3–4%.20,21 La adición de quimioterapia incrementa la vida media en forma notable,22 el metotrexato ha demostrado tener un mayor efecto. Sin embargo, el principal problema relacionado con la administración de dicho medicamento en estos pacientes, es la neurotoxicidad, misma que se potencia por la aplicación de la radioterapia. La posibilidad de neurotoxicidad asociada al metotrexato no es despreciable, dado que se ha reportado con una frecuencia de 13 a 26% y se incrementa notablemente en pacientes mayores de 60 años.23,24 A pesar de ello, aun con el riesgo de neurotoxicidad, el manejo actual más aceptado para el LPSNC en pacientes IC es la terapia combinada, que ofrece supervivencia de 17–44 meses con un porcentaje que oscila entre los 25 a 45% de supervivencia a cinco años con una aceptable calidad.6
En el presente trabajo hemos seleccionado a los pacientes con LPSNC en quienes se descartaron síndromes de inmunodeficiencia, a fin de realizar un análisis objetivo de las manifestaciones clínicas, comportamiento radiológico, aspecto histopatológico, dificultades diagnósticas y terapéuticas, así como consideraciones pronósticas.
Pacientes y métodos
Se realizó el análisis retrospectivo de pacientes inmuno–competentes con lesiones cerebrales diagnosticadas como LPSNC atendidos en el Servicio de Neurocirugía del Hospital de Especialidades del Centro Médico Nacional Siglo XXI durante un período de 10 años. En todos los casos se realizó un análisis clínico donde se consideró: la edad, sexo, antecedentes patológicos, en especial neoplásicos, signos y síntomas característicos, así como las medidas terapéuticas utilizadas. En todos se descartaron, tanto linfomas en localización diferente al sistema nervioso central, como enfermedades por inmunodeficiencia, en especial SIDA. Se realizaron estudios de ecografía, tomografía computada abdominal, tomografía torácica, resonancia magnética (RM) y pruebas inmunológicas específicas para descartar la presencia del virus de inmunodeficiencia humana (VIH), enfermedades autoinmunes, así como infecciones por otros virus (hepatitis C y HTLV) o Helicobacter pylori.
En el presente estudio se incluyeron 22 pacientes quienes cumplieron con todos los criterios antes descritos. Los pacientes fueron ingresados con diagnóstico de lesión o lesiones cerebrales en estudio y todos fueron sometidos inicial–mente a manejo con esteroides por vía intravenosa y toma de biopsia por estereotaxia. Los tejidos fueron teñidos con hematoxilina y eosina o preparados para inmunohistoquímica para su estudio con marcadores específicos para linfocitos B y T.
Una vez confirmado el diagnóstico y basados en previos informes,12,15,18,22,25 a todos los pacientes se les planteó la opción terapéutica de quimioterapia inicial con metotrexato y radioterapia, proporcionándoles una explicación amplia con respecto a la supervivencia, así como de los posibles efectos adversos. Diez de los 22 pacientes aceptaron el tratamiento combinado con quimioterapia y radioterapia, mientras que el resto fue sometido únicamente a radioterapia. Para la radioterapia se administraron dosis totales de 40 Gy con un promedio de 20 sesiones, llevándose a cabo cada dos días, y dividiéndose el encéfalo en regiones laterales: derecha e izquierda. El metotrexato fue aplicado a dosis de 3.5 g/m2/ semana cada dos semanas por 6 sesiones, buscándose dosis citotóxicas para células de linfoma con una concentración mayor a 1 µmol/dL.
El período de seguimiento fue de 3 a 72 meses, tiempo en el que, además de la valoración clínica con visitas hospitalarias cada mes y medio, se efectuaron estudios de imagen como control, en especial resonancia magnética de encéfalo simple y con medio de contraste cada tres meses, como parte del protocolo, a fin de detectar la posible recurrencia tumoral, y en especial cambios sugestivos de radionecrosis.
Resultados
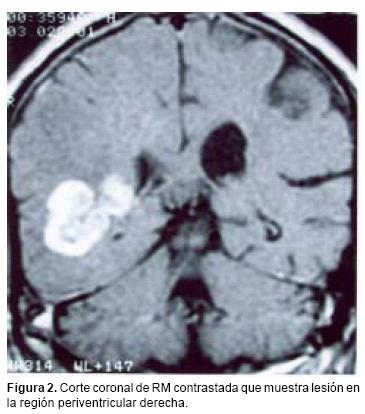
La distribución por sexo de los 22 pacientes, fue equitativa (Cuadro I) con un promedio de edad de 65 años (límites entre 43 y 92 años). Cinco pacientes refirieron antecedentes neoplásicos heredofamiliares en rama directa: dos carcinomas cervicouterinos, dos adenocarcinomas de pulmón y un carcinoma de células gigantes. En un solo paciente se encontró antecedentes personales de neoplasia, por melanoma diagnosticado 10 años antes. El tiempo promedio entre la aparición del primer síntoma y la hospitalización fue de 80 días (límites entre 7 y 730), el cual fue con mayor frecuencia cefalea asociada a déficit neurológico focal. Un paciente presentó síntomas inespecíficos, tales como febrícula y alteraciones gastrointestinales antes del inicio del cuadro clínico, sin asociación al padecimiento neurológico. El promedio de calidad de vida de acuerdo a la escala de Karnofsky26 fue de 62.7 años. Los estudios radiológicos mostraron en cuatro pacientes lesiones múltiples, mientras que en el resto fueron lesiones únicas. La localización de cada lesión fue como sigue: 21 en los hemisferios cerebrales (ocho parietales, seis frontales, cuatro temporales y tres occipitales), seis en los núcleos basales, cuatro en el cuerpo calloso y uno en el tallo cerebral. En la tomografía computada las lesiones se apreciaron isodensas con discreta tendencia a la hiperdensidad y con un reforzamiento regular y marcado a la administración del medio de contraste (Figura 1). En la resonancia magnética, las lesiones aparecieron como iso o discretamente hipointensas en T1, con evidente y homogéneo reforzamiento con la aplicación del medio de contraste (Figura 2); en la fase T2 la mayoría de las lesiones eran hiperintensas, acompañándose de un marcado edema circundante. El tamaño promedio de los tumores fue de 22 mm (límites entre 3 y 57 mm), localizándose la mayoría de ellas en la región periventricular o subcortical, adyacentes a la circulación del líquido cefalorraquídeo; en el cuerpo calloso se ubicaban principalmente en el esplenio (Figura 3).


Los resultados de las biopsias realizadas por estereotaxia, mostraron que sólo en tres pacientes el diagnóstico histológico transoperatorio correspondió a linfoma, y en el resto fue confundido con astrocitomas, metástasis o glioblastomas multiformes. En el análisis de las biopsias por microscopia de luz a menor aumento fue posible observar un patrón de infiltración angiocéntrico, formando collares de células tumorales rodeando pequeños vasos cerebrales (Figura 4). Con mayor aumento fue posible observar con más detalle el cúmulo de células, en donde en algunos casos consistió en una combinación de células grandes y pequeñas pero persistiendo el ordenamiento perivascular (Figura 5). Sin embargo, en los pacientes con estadios más avanzados de la enfermedad se apreció pérdida del ordenamiento perivascular, con invasión de las células tumorales al parénquima cerebral. También se observó reacción astrocítica asociada, además de la presencia de macrófagos. La mayoría de los tumores fueron considerados de la variedad de células grandes difusas, de acuerdo a la Clasificación Revisada Europea–Americana de Linfoma (REAL),27 y en un solo caso correspondió a la variedad anaplásica. Por inmuhistoquímica, el total de los tumores resultaron positivos a los antígenos CD20 y CD79A, por lo que se consideraron de células B.

La supervivencia promedio en 12 pacientes tratados únicamente con radioterapia fue de 11 meses, la que se incrementó a 36 meses en los 10 pacientes restantes que recibieron además tratamiento con metotrexato. En la mayoría de los pacientes sometidos al tratamiento combinado (radio y quimioterapia) se notó franca mejoría no sólo en el cuadro clínico, sino también en los estudios de imagen realizados durante el seguimiento, apreciándose incluso, la desaparición de la lesión o lesiones (Figura 6). Todos los pacientes que recibieron quimioterapia presentaron alteraciones en las pruebas de funcionamiento hepático, sin llegar al daño hepático agudo; además, dos de ellos mostraron datos de insuficiencia renal aguda con duración de 10 días en promedio. En un caso se presentó un cuadro de gastritis aguda requiriendo de su hospitalización y tratamiento intravenoso con inhibidores de bomba de protones. Los pacientes que recibieron el tratamiento combinado presentaron astenia y adinamia intensa mientras se aplicó el tratamiento.

El tiempo de recurrencia tu moral fue notablemente menor en los pacientes tratados sólo con radioterapia, la que fue en promedio de seis meses, mientras que los sometidos a la terapia combinada fue de 24 meses; los pacientes que presentaron recurrencias se manejaron solamente en forma conservadora a base de anticonvulsivos y esteroides para el control del edema cerebral. Las lesiones recurrieron en el mismo sitio en 12 de los 22 casos, mientras que en cinco se presentaron en otros sitios; los cinco pacientes restantes fallecieron antes de mostrar imágenes radiográficas sugestivas de recurrencia. Dos de estos pacientes fallecieron en forma súbita en su domicilio por causas desconocidas, uno más presentó hemorragia cerebral relacionada con hipertensión arterial; finalmente, dos pacientes que habían sido sometidos a radio y quimioterapia presentaron ataxia y síndrome demencial progresivo que les afectó notablemente su calidad de vida, falleciendo al poco tiempo de haber culminado el tratamiento. A uno de ellos fue posible realizarle resonancia magnética, días antes del fallecimiento, la que mostró imagen hipointensa difusa subcortical con reforza–miento heterogéneo a la aplicación del medio de contraste, muy sugestiva de leucoencefalopatía (Figura 7). El mayor tiempo de supervivencia se encontró en aquellos pacientes que presentaron menor déficit neurológico al momento del diagnóstico, así como menor edad y mayor tiempo de evolución del cuadro clínico.

Discusión
Si consideramos que durante el período del presente estudio se intervinieron quirúrgicamente un total de 6,357 pacientes con tumores cerebrales en el Servicio de Neurocirugía del Hospital de Especialidades del Centro Médico Nacional Siglo XXI, la frecuencia de aparición en nuestra población de LPSNC en pacientes IC fue de 0.34%, situación que lo hace un tumor poco frecuente.
El promedio de edad referido en las publicaciones similares a la presente es de 61 años,28 lo que coincide con nuestros hallazgos. Asimismo, al igual que lo informado, no encontramos diferencia estadística con respecto al sexo y la frecuencia de aparición de esta enfermedad. Dentro de los antecedentes heredofamiliares, se ha mencionado que cuando los pacientes con LPSNC presentan historia familiar de adenocarci–nomas o leucemias el pronóstico tiende a ser peor,9 aspecto que no se corroboró en este estudio.
El tiempo promedio entre la aparición del primer síntoma y la hospitalización estuvo de acuerdo con lo publicado por otras series.29 Si bien en un paciente se presentaron síntomas generales que pudieran considerarse como "síntomas B", en realidad no hubo relación alguna con la enfermedad neurológica, por lo que no se tomó en cuenta al momento de realizar el análisis.
Como fue posible observar, todos los pacientes de nuestro grupo recibieron esteroides antes de ser sometidos a la biopsia por estereotaxia. Este hecho no fue parte del protocolo; sino obedeció al manejo del edema con la finalidad de atenuar las manifestaciones clínicas. Esta observación es importante ya que los esteroides pueden ocasionar la desaparición de las lesiones, motivo por el cual al LPSNC también se le conoce con el nombre de "tumor fantasma".30,31 Es importante hacer notar que en estos casos, el tumor recurre invariablemente unos meses más tarde, siendo refractario a un segundo tratamiento. Este dato también puede explicar la dificultad diagnóstica transoperatoria en nuestros casos, dado el cambio en la citoarquitectura que los esteroides ocasionan en estos tumores. En la actualidad, ante la sospecha de un linfoma primario no se recomienda el uso de esteroides como primera intención, sino después de haber realizado la toma de biopsia, o bien administrarlo sólo en aquellos casos donde el edema cerebral sea tan marcado que ponga en peligro la integridad neurológica del paciente. Por otro lado, el manejo con esteroides de ninguna manera deberá utilizarse como "prueba terapéutica", dado que existen otras lesiones como la sarcoidosis o la esclerosis múltiple, que son radiológicamente similares al LPSNC y que también pueden responder a este régimen. Una vez establecido el diagnóstico, sí se recomiendan estos medicamentos tanto por su efecto citotóxico en el tumor, como para el control del edema cerebral.
En nuestros pacientes observamos multiplicidad de lesiones con frecuencia mayor a lo informado por Bühring y cols;32 sin embargo, su patrón de distribución en cuanto a la localización en el encéfalo fue muy similar. Además, contrario a lo informado por Namasivayam y Teasdale,33 no hubo repercusión entre la multiplicidad de las lesiones y el pronóstico clínico. Los hallazgos clínicos que tuvieron un impacto en la supervivencia fueron: la edad, la calidad de vida previa a la intervención, así como el tiempo de evolución del padecimiento. Estas observaciones están de acuerdo con Kim y Ferrei,34,35 quienes refieren un mal pronóstico en pacientes mayores de 60 años, con una puntuación en la escala de Karnofsky menor de 70 y duración de los síntomas menor a cuatro semanas. Balmaceda y cols,36 informan que la invasión leptomeníngea del tumor es uno de los principales indicadores de mal pronóstico, hecho que en ocasiones es muy difícil de demostrar. El diagnóstico se basa en hallazgos por resonancia magnética de reforzamiento leptomeníngeo, así como por análisis citológicos en el líquido cefalorraquídeo y/o la elevación de la isoenzima 5 de la deshidrogenasa láctica, hallazgos que son poco específicos, por lo que la validez estadística de este dato es muy cuestionable.
Los hallazgos por resonancia magnética concuerdan con los informados en otros estudios.37–39 En primer lugar, el tamaño promedio de las lesiones coincidió con los 19.9 mm informados. Pero lo más significativo de las imágenes radiográficas fue que, basado en estos hallazgos y comparándolos con lo publicado, se pudo establecer diferencias entre la descripción radiológica clásica del LPSNC informados en pacientes VIH–positivos con respecto a los que presentaron los pacientes IC. Esta diferencia consistió en el reforzamiento con el medio de contraste, debido a que en los pacientes con SIDA se ha informado que éste tiende a presentar una morfología anular, mientras que en los VIH–negativos la distribución del medio de contraste es más homogénea en toda la lesión, hecho que pudo ser corroborado en nuestro trabajo. Finalmente, contrario a lo informado por Johnson y cols,38 no se encontró correlación entre el grado de necrosis en el estudio histopatológico y la intensidad en la fase T2 en la resonancia magnética.
Los hallazgos histopatológicos de los tumores fueron iguales a los clásicamente reportados en estudios similares,14–16 así como los estudios de inmunohistoquímica,40,41 donde se corroboró que la totalidad de los tumores estaban formados por linfocitos B. Aunque hay informes de tumores formados por linfocitos T, en realidad esta diferenciación es solamente estructural, dado que no se ha encontrado una diferencia clínica estadísticamente significativa entre uno y otro grupo (T y B).42 Por otro lado, es bien conocida la existencia de varias y complejas clasificaciones histológicas para los LPSNC; nosotros utilizamos la REAL por ser la más práctica y conocida en la literatura internacional. Sin embargo, todas estas clasificaciones son solamente de utilidad morfológica, dado que tampoco existe diferencia clínica alguna entre los distintos grupos en cada una de ellas.6
Con base en informes previos,22 nuestro protocolo de manejo incluyó la toma de biopsia por estereotaxia, quimioterapia con metotrexato y radioterapia. Una vez que se plantearon los riesgos de este régimen, solamente 10 pacientes aceptaron completarlo, el resto de ellos desistió al conocer los posibles efectos secundarios. Sin embargo, fue posible observar un mayor tiempo de supervivencia sin afectar la calidad de vida, precisamente en estos 10 pacientes en quienes se utilizó el tratamiento completo. Aun cuando dos de estos pacientes presentaron signos clínicos y radiológicos de neurotoxicidad, al analizar en forma global los resultados, se corroboró lo que en otras publicaciones en el sentido de que esta es la mejor alternativa terapéutica,6 dado que se pudo incrementar de 11 a 36 meses en promedio el tiempo de supervivencia con una aceptable calidad. Los efectos adversos que mayormente se han informado con el uso de metotrexato asociado a radioterapia son: neurotoxicidad, mucositis, falla hepática y falla renal aguda. El metotrexato se excreta primariamente en el riñón, dificultando su indicación en pacientes con antecedentes de problemas renales. Una vez utilizado, es necesario mantener un pH mayor a 7.0 porque se ha visto que cuando el pH es ácido, puede ocasionar precipitación del metotrexato en los túbulos proximales renales causando falla renal aguda. En dos de nuestros pacientes se encontró esta complicación, misma que se trató adecuadamente y sin consecuencias.
Considerando todos estos efectos adversos de la terapia combinada, es indispensable plantear claramente esta situación y que, en forma conjunta, el enfermo, los familiares y el médico tomen la decisión que se considere más conveniente, acorde a las condiciones propias de cada caso.
Referencias
1.Bailey P. Intracranial sarcomatous tumors of leptomeningeal origin. Arch Surg 1929; 18:1359–1402. [ Links ]
2. Henry JM, Heffiner RRJ, Drillard SH Earle KM, Davis RL. Primary malignant lymphomas of the central nervous system. Cancer 1974; 34:1293–1302. [ Links ]
3. Guinto G, Félix I, Aréchiga N, Arteaga V, Kovacs K. Primary central nervous system lymphomas in immunocompetent patients. Histol Histopathol 2004; 19:963–972. [ Links ]
4. DeAngelis LM. Primary central nervous system lymphoma. J Neurol Neurosurg Psychiatry 1999; 66:699–701. [ Links ]
5. Postler E, Bornemann A, Skalej M, Krober S, Kaiserling E, Wickboldt J, Meyermann R. Intracranial inflammatory tumors: a survey of their various etiologies by presentation of 5 cases. J Neurooncol 1999; 43:209–217. [ Links ]
6. Paulus W, Jellinger K, Morgello S, Dekert–Schlüter. Malignant lymphomas. En: Pathology and genetics of tumours of the nervous system. Kleihves P, Cavenee WK (Eds). International Agency for Reserch on Cancer. Lyon, France 2000. p. 198–203. [ Links ]
7. Basso U, Brandes A. Diagnostic advances and new trend of the treatment of primary central nervous system lymphoma. Eur J Cancer 2002; 38:1298–1312. [ Links ]
8. Kadan–Lottick NS, Skluzacek MC, Gurney JG. Decreasing incidence rates of primary central nervous system lymphoma. Cancer 2002; 95:193–202. [ Links ]
9. Bataille B, Delwail V, Menet E, Vandermarcq P, Ingrand P, Wager M, Guy G, Lapierre F. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg 2000; 92:261–266. [ Links ]
10. Geddes JF, Bhattacharjee MB, Savage K, Scaravalli F, McLaughlin JE. Primary cerebral lymphoma: a study of 47 cases probed for Epstein–Barr virus genome. J Clin Pathol 1992; 45: 587–590. [ Links ]
11. Chang KL, Flaris N, Hickey WF, Johnson RM, Meyer JS, Weiss LM. Brain lymphomas of immunocompetent and immunocompromised patients: study of the association with Epstein–Barr virus. Mod Pathol 1993; 6:427–432. [ Links ]
12. Remick SC, Diamond C, Migliozzi JA, Solis O, Wagner H, Haase RF, et al. Primary central nervous system lymphoma in patients with and without acquired immune deficiency syndrome. A retrospective analysis and review of the literature. Medicine (Baltimore) 1990; 69:345–360. [ Links ]
13. Jellinger KA, Paulus W. Primary central nervous system lymphomas–an update. J Cancer Res Clin Oncol 1992; 119:7–27. [ Links ]
14.Adams J, Howatson AG. Cerebral lymphomas: review of 70 cases. J Clin Pathol 1990; 43:544–547. [ Links ]
15. O'Neill BP, Kelly PJ, Earle JD, Scheithauer B, Banks PM. Computer assisted stereotaxic biopsy for the diagnosis of primary cent ral nervous system lymphoma. Neurology 1987; 37:1160–1164. [ Links ]
16. Ashby MA, Bowen D, Bleehen NM, Barber PC, Freer CE. Primary lymphoma of the cent ral nervous system: experience at Addenbrooke's Hospital, Cam bridge. Clin Radiol1 988; 39:173–181. [ Links ]
17. Kepes JJ. Astrocytomas: old and newly recognized variants, their spectrum and morphology and antigen expression. Can J Neurol Sci 1987; 14:109–121. [ Links ]
18. Murray K, Kun L, Cox J. Primary malignant lymphoma of the central nervous system. Results of treatment of 11 cases and review of the literature. J Neurosurg 1986; 65:600–607. [ Links ]
19. O'Niell BP, Illig J J. Primary central nervous system lymphoma. Mayo Clin Proc 1989; 64: 1005–1020. [ Links ]
20. Nelson DF, Martz KL, Bonner H, Nelson JS, Newall J, Kerman HD, et al. Non–Hodgkin's lymphoma of the brain: can high dose, large volume irradiation therapy improve survival? Report on a prospective trial by the Radiation Therapy Oncology Group (RTOG): RTOG 8315. Int J Radiat Oncol Biol Phys 1992; 23:9–17. [ Links ]
21. Blay JY, Ongolo–Zogo C, Sebban C, Thiesse P, Biron P. Primary cerebral lymphomas: unsolved issues regarding first line treatment, follow–up, late neurological toxicity and treatment of relapses. Ann Oncol 2000; 11:39–44. [ Links ]
22. DeAngelis LM, Yahalom J, Thaler HT, Kher U. Combined modality therapy for primary CNS lymphoma. J Clin Oncol 1992; 10:635–643. [ Links ]
23. Abrey LE, DeAngelis LM, Yahalom J. Long–term survival in primary CNS lymphoma. J Clin Oncol 1998; 16:859–863 [ Links ]
24. Blay JY, Conroy T, Chevreau C, Thyss A, Quesnel N, Eghbali H, et al. High dose methotrexate for the treatment of primary central nervous system lymphomas: analysis of survival and late neurological toxicity in a retrospective series. J Clin Oncol 1998; 16:864–871. [ Links ]
25. Nasir S, DeAngelis LM. Update on the management of primary CNS lymphoma. Oncology (Williston Park) 2000; 14:228–234. [ Links ]
26. Karnofsky DA, Abelman WH, Craver LF, Burchenal JH. The use of the nitrogen mustards in the palliative treatment of carcinoma with particular reference to bronchogenic carcinoma. Cancer 1948; 1:634–656. [ Links ]
27. Harris Nl, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML,et al. A revised European–American Classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994; 84:1361–1392. [ Links ]
28. Tomlinson FH, Kurtin PJ, Suman VJ, Scheithauer BW, O'Fallon JR, Kelly PJ, et al. Primary intracerebral malignant lymphoma: a clinicopathological study of 89 patients. J Neurosurg 1995; 82:558–566. [ Links ]
29. Hayabuchi N, Shibamoto Y, Onizuka Y. Primary central nervous system lymphoma in Japan: a nationwide survey. Int J Radiat Oncol Biol Phys 1999;44:265–272. [ Links ]
30. Pirotte B, Levevier M, Golmann S, Brucher JM, Brotchi J, Hildebrand J. Glucocorticoid–induced long term remission in primary cerebral lymphoma: case report and review of the literature. J Neurooncol 1997; 32:63–69. [ Links ]
31. Filla A, De Michele G, D'Armiento FP, Di Salle F, Cirillo S, Marano E, et al. Steroid–induced disappearance of primary central nervous system lymphoma. Clinical, neuroradiological and pathological findings. Acta Neurol (Napoli) 41. 1989; 11:400–407. [ Links ]
32. Bühring U, Herrlinger U, Krings T, Thiex R, Weller M, Küker W. MRI features of primary central nervous system lymphomas at presentation. Neurology 2001; 57:393–396. [ Links ]
33. Namasivayam J, Teasdale E. The prognostic importance of CT features in primary intracranial lymphoma. Br J Radiol 1992; 65:761–765. [ Links ]
34. Kim DG, Nam DH, Jung HW, Choi KS, Han DH. Primary central nervous system lymphoma: variety of clinical manifestations and survival. Acta Neurochir (Wien) 1996; 138:280–289. [ Links ]
35. Ferreri AJ, Blay JY, Reni M, Pasini F, Spina M, Ambrosetti A, et al. Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol 2003; 15:266–272. [ Links ]
36. Balmaceda C, Gaynor JJ, Sun M, Gluck JT, DeAngelis LM. Leptomeningeal tumor in primary central nervous system lymphoma: recognition, significance and implications. Ann Neurol 1995; 38:202–209. [ Links ]
37. Watanabe M, Tanaka R, Takeda N, Wakabayashy K, Takahashi H. Correlation of computed tomography with the histology of primary malignant lymphoma of the brain. Neuroradiology 1992; 34:36–42. [ Links ]
38. Johnson BA, Fram EK, Johnson PC, Jacobowitz R. The variable MR appearance of primary lymphoma of the central nervous system: comparison with histopathologic features. Am J Neuroradiol 1997; 18:563–572. [ Links ]
39. Erdag N, Bhorade RM, Alberico RA, Yousuf N, Patel MR. Primary lymphoma of the central nervous system: Typical and atypical CT and MR imaging appearances. Am J Roentgenol 2001; 176:1319–1326. [ Links ]
40. Paulus W, Jellinger K. Comparison of integrin adhesion molecules expressed by primary brain lymphomas and nodal lymphomas. Acta Neuropathol (Berl)1993; 86:360–364. [ Links ]
41. Murphy JK, O'Brien CJ, Ironside JW. Morphologic and immunophenotypic characterization of primary brain lymphomas using paraffin–embedded tissue. Histopathology 1989; 15:449–460. [ Links ]
42. McCue MP, Sandrock AW, Lee JM, Harris NL, Hedley–Whyte ET. Primary T– cell lymphoma of the brainstem. Neurology 1993; 43:377–381. [ Links ]

