










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkGaceta médica de México
versão On-line ISSN 2696-1288versão impressa ISSN 0016-3813
Gac. Méd. Méx vol.141 no.3 Ciudad de México Mai./Jun. 2005
Ejercicio clínico patológico
Mujer de 42 años con ascitis y presencia de linfadenopatía generalizada
A 42 year–old woman with ascitis and presence of generalized linfadenopathy
Senen Lopéz–Krauletz,ª* Haiko Nellen–Hummel,ª Andrés Jacobo–Rubalcava,ª Verónica Iparraguirre–Palomeque,ª Fernando Flores–Figueroa,ª Ismael Ayala–Hernández,ª José Halabe–Cheremª
ª Departamento de Medicina Interna, Hospital de Especialidades del Centro Médico Nacional Siglo XXI, IMSS, México, D.F., México
Hospital de Especialidades del Centro Médico Nacional Siglo XXI, IMSS.
Coordinador: Manuel de la Llata–Romero
Colaboradores: Juan Urrusti–Sanz
Jesús Aguirre– García Roberto Sánchez–Ramírez
* Correspondencia y solicitud de sobretiros:
Senen López–Krauletz.
Departamento de Medicina Interna. Hospital de Especialidades del Centro Médico Nacional Siglo XXI. IMSS. Av. Cuauhtémoc No. 330, Col. Doctores,
C.P. 06700 México, D.F.
Presentación del caso
Mujer de 42 años de edad, originaria y residente del estado de Chiapas. En febrero del 2001 presentó disnea, astenia, adinamia, dolor torácico inespecífico, fiebre, hematuria, lumbalgia, ascitis y cursó con un cuadro de oclusión intestinal que ameritó laparotomía exploradora en la cual no se encontraron alteraciones. Después de la cirugía presentó deterioro de su estado general, requiriendo asistencia mecánica ventilatoria durante 18 días; cursó con deterioro de la función renal, elevación de azoados, oliguria y albuminuria, así como falla orgánica múltiple. Su evolución tendió a la mejoría, sin embargo, presentó retención de líquidos, derrame pleural y pericárdico de 150mL, ascitis y hematuria. Se realizó USG abdominal por el que se descartó cirrosis hepática e hipertensión portal, y los riñones se encontraron de tamaño normal. Se manejó con diuréticos y medidas sintomáticas, evolucionando a la mejoría y egresando asintomática.
En agosto del 2003 presentó nuevamente ascitis a tensión sin otra sintomatología. La exploración física reveló ligero tinte ictérico, en cuello se palparon pequeñas adenomegalias múltiples en la región submaxilar bilateral, submentoniana, adenomegalias pre y retroauriculares, cervicales anteriores y posteriores. Los pulmones con disminución del murmullo vesicular y vibraciones vocales en región basal derecha, a la percusión submatidez sugerente de derrame pleural mínimo, aproximadamente 10 a 15%. El abdomen se encontró con aumento de volumen debido a líquido de ascitis, sin red venosa colateral, la peristalsis era normal, tenía adenomegalias en regiones inguinales, no dolorosas ni fijas a tejidos profundos, las extremidades sin edema.
En los estudios efectuados se encontró: Urea, 218 mg/dL; BUN, 101.8 mg/dL; Creatinina, 4.3 mg/dL; albúmina, 2.1 g/dL; potasio, 4.0 mEq/L; sodio, 133 mEq/L; Hb, 7.9 g/dL; leucocitos, 29.1x103/mm3; plaquetas, 590x103/mm3.
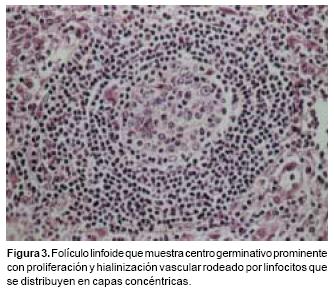
Requirió transfusión de concentrados eritrocitarios y plaquetarios. La B2–microglobulina fue de 4814 mg/dL. La radiografía de tórax mostró ensanchamiento del mediastino, una tomografía de tórax evidenció tumoración en mediastino anterior (Figura 1) y en el ultrasonido pélvico había líquido libre en cavidad con útero y anexos normales (Figura 2). El aspirado de médula ósea reveló cambios megaloblásticos y daño tóxico medular. Biopsia de hueso normal. En ascitis se determinó adenosindeaminasa y BAAR, así como Papanicolaou, con resultados negativos. Se realizó biopsia excisional de adenomegalias cervicales reportando hiperplasia angiofolicular (Enfermedad de Castleman de tipo Hialino–Vascular) sistémica, (Figura 3).


Comentario clínico
La enfermedad de Castleman, descrita en 1956, también llamada hiperplasia linfoide angiofolicular, consiste en proliferación masiva del tejido linfoide.1
Se han descrito tres variantes histológicas, la hialino vascular, de células plasmáticas y la mixta. Clínicamente puede presentarse en forma localizada en la que el 80% de su estirpe histológica es hialino vascular y cerca de 20% de células plasmáticas y ocasionalmente mixtas. La variedad de células plasmáticas es más común en niños y jóvenes, y se presenta como tumores mediastinales y abdominales, raramente adenopatías periféricas, cursa con anemia, hipoalbuminemia, hipergammaglobulinemia, velocidad de sedimentación globular elevada, hemoptisis, pérdida de peso y fiebre.1–3
La forma sistémica generalmente es de células plasmáticas, aunque ocasionalmente puede ser hialino vascular o mixta con menor frecuencia. A diferencia de la enfermedad localizada cursa con linfadenopatía generalizada y tiene un curso agresivo, usualmente fatal, puede cursar con complicaciones infecciosas y con riesgo de transformación tumoral, como linfoma y sarcoma de Kaposi.
Se han encontrado secuencias de DNA del herpesvirus tipo 8, que contienen un homólogo de la IL–6 humana en su genoma que estimula y actúa como un factor de crecimiento en diversas entidades, incluyendo el sarcoma de Kaposi, la enfermedad de Castleman sistémica y el mieloma múltiple, asociando al síndrome de POEMS con el mieloma otoesclerótico en 90% de los casos y en la enfermedad de Castleman generalizada en 60% de los casos.1,4,6,7,9 En este caso, la paciente cursó con datos clínicos para POEMS incompleto. En ocasiones se asocia a amiloidosis renal tipo AA pudiendo ésta cursar con síndrome nefrótico y falla renal.pdf elaborado en medigraphic8 En nuestro caso ocurrió esto.
En la enfermedad de Castleman localizada, el tratamiento es quirúrgico, demostrando ser efectivo y curar la enfermedad sin evidencia de recurrencia, con un buen pronóstico; sin embargo en casos de cirugía incompleta existe el riesgo de recidiva.1,2 En estos pacientes es conveniente realizar la linfocentellografía con galio 67 en busca de ganglio centinela para normar la conducta quirúrgica, ya que existen informes de recaídas de la enfermedad nueve años después del diagnóstico inicial.5
En la enfermedad generalizada o sistémica, la conducta terapéutica es más controversial, algunos autores manejan únicamente medidas generales de acuerdo al estado clínico del paciente, a la variedad histológica, a la afectación de diferentes órganos, a las alteraciones en parámetros de laboratorio, a la afectación del mediastino y a la presencia de ascitis. En función a estos resultados se han utilizado diversos esquemas de quimioterapia con respuestas hasta de remisión completa. Sin embargo existen casos de recidiva, requiriendo en la mayoría de los esquemas tratamiento de sostén mediante esteroides a dosis de 20–30mg/día de prednisona, con el riesgo de mayor daño al sistema inmunológico, ya de por sí deteriorado en estos pacientes y, las complicaciones infecciosas siendo una causa importante de muerte en este grupo de pacientes. Así mismo se ha obtenido respuesta parcial a la radioterapia, e incluso a quimioterapia más cirugía en algunos casos. Sin embargo, la transformación a sarcoma de Kaposi, o a linfoma de células T, desde los 10 meses hasta los 13 años, con un pronóstico fatal, sobre todo cuando se asocia a variedad de células plasmáticas, teniendo un pronóstico más favorable en la enfermedad sistémica, variedad hialinovascular, como nuestra paciente.1,2 La paciente que nos ocupa hasta el momento de esta publicación se encuentra asintomática sin datos de enfermedad activa. Su control en la consulta externa deberá prolongarse durante varios años.
Referencias
1. Herrada J, Cabanillas F, Rice L, et al. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med 1998; 128(8):657–662. [ Links ]
2. Frizzera G, Bayrd E, Goldman A. A systemic lymphoproliferative disorder with morphologic features of Castleman's disease: clinical findings and clinicopathologic corelations in 15 patients. JCO 1985; 3(9):1202–1216. [ Links ]
3. Ahmed R, Adhami N, Hanif M, et al. Unicentric Castleman disease; a case report and review of literature. Chest 1999; 116(4):410S. [ Links ]
4. Maslovsky I, Uriev L, Lugassy G. The heterogeneity of Castleman disease: Report of five cases and review of literature. Am J Med Scien 2000; 320(4):292–295. [ Links ]
5. Kinoshita T, Ishii K, Mori Y, et al. Castleman disease in the anterior neck: the role of Ga–67 scintigraphy. Clin Nucl Med 1996; 21(8):626–628. [ Links ]
6. Belec L, Hallouin M, Authier F, et al. Human herpesvirus 8 DNA sequences with restricted variability in ORF26 in patients with POEMS syndrome–asociated Castleman's disease. Neurology 1998; 504(4):A184S. [ Links ]
7. Zeidman A, Fradin Z, Cohen A, et al. Kaposi's sarcoma associated with Castleman's disease. Eur J Haematol 1999; 63(1):67–70. [ Links ]
8. Kazes I, Deray G, Jacobs C. Castleman disease and renal amyloidosis. Ann Intern Med 1995; 122(5):395–396. [ Links ]
9. Norihiro N, Mitsuko S, Yoshihito S, et al. Improvement in Castleman's disease by humanized anti–interleukin–6 receptor antibody theraphy. Blood 2000; 95(1):56–61. [ Links ]

