










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkGaceta médica de México
On-line version ISSN 2696-1288Print version ISSN 0016-3813
Gac. Méd. Méx vol.140 n.5 Ciudad de México Sep./Oct. 2004
Ejercicio clínico patológico
Hospital de Especialidades del Centro Médico Nacional Siglo XXI, IMSS
Hombre de 35 años de edad con defecto campimétrico y alteraciones medulares
A Thirty Five Years Old Male With Campimetric Defect and Molecular Alterations
Carlos Cuevas–García,* Selene Vega–Gaxiola,** Cristina Rivera–Nava,*** María Luisa Márquez–Rocha****
* Jefe del servicio de Neurología, Hospital de Especialidades del Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social.
** Residente de Tercer año del servicb de Neurología.
*** Médico adscrito al Servicio de Neurología
**** Médico adscrito al Servicio de Neuropatología. Hospital de Especialidades del Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social.
Coordinador: Dr. Manuel de la Llata–Romero
Colaboradores: Dr. Juan Urrusti–Sanz
Dr. Jesús Aguirre–García
Dr. Roberto Sánchez–Ramírez
Correspondencia y solicitud de sobretiros:
Océano 83, Jardines del Pedregal,
Del. Alvaro Obregón,
C.P. 01900, México, D.F.
correo electrónico: cacu@avantel.net.
Resumen clínico
Hombre de 35 años de edad, originario y residente de México DF. Casado, católico, escolaridad secundaria, ocupación obrero, nivel socioeconómico bajo.
Antecedentes personales no patológicos. Tabaquismo positivo en los últimos 15 años a razón de 10 unidades por día.
Antecedentes personales patológicos: relaciones sexuales sin uso de condón.
Padecimiento actual: inicia 15 días previos a su ingreso con fiebre, rinorrea verde, malestar general, mialgias, artralgias, cefalea holocraneana pulsátil, de intensidad moderada , que se exacerbaba con la tos y los cambios de posición, pero que le permitía continuar con sus actividades diarias y mejoraba poco mediante analgésicos comunes; recibió Penicilina durante 4 días sin mejoría de los síntomas generales.
Siete días después evoluciona refiriendo sensación de cosquilleo en manos y sensación de prurito y adormecimiento en los muslos, así como persistencia de la cefalea en forma intermitente. Dos días después, la cefalea aumenta de intensidad, sin nausea ni vómito pero con visión borrosa y notan que empieza a tropezar con los objetos del lado izquierdo de su campo visual.
Finalmente es internado en este Hospital por presentar, además de las manifestaciones previas, desorientación, olvidos frecuentes, labilidad emocional , mayor hipoestesia de miembros inferiores, retención aguda de orina, constipación intestinal e incapacidad para movilizar las extremidades inferiores.
Exploración clínica
Frecuencia cardiaca 80x', tensión arterial 130/80 mmHg, frecuencia respiratoria 18x', temperatura 37°C
Funciones mentales: alerta pero desorientado en lugar, tiempo y persona; inatención en forma intermitente, lenguaje adecuado, lateralidad diestra.
A la exploración física general, talla baja, palidez de piel y mucosas, buena hidratación y estado general regular. Se apreció hiperemia faríngea con exudado blanco sobre paladar blando. Tórax en tonel, campos pulmonares bien ventilados. Corazón con ruidos cardiacos rítmicos sin soplos u otras alteraciones. Abdomen blando, depresible, no dobroso, sin visceromegalias. Extremidades con pulsos presentes y palpables.
Examen neurológico: nervios craneales: I diferido. II el fondo de ojo y colorimetría normales; campimetría por confrontación con hemianopsia homónima izquierda. Nervios craneales III, IV y VI, normales incluyendo reflejos pupilares directos, consensúales y de acomodación. La exploración de mirada conjugada bilateral demostró sacadas hipométricas bilaterales y nistagmus optoquinético disminuido de derecha a izquierda. El resto de los nervios craneales explorados fueron normales.
Fuerza muscular: Miembros superiores 5/5; Miembros inferiores 0/5 (paraplejia). Reflejos de estiramiento muscular ++/++++ en miembros superiores y 0/++++ en miembros inferiores (arreflexia).
Sensibilidad: abolida en todas sus modalidades (exterocepción y propiocepción) desde el nivel de T7.
Había ausencia de reflejos abdominocutáneos superior e inferior y cremasteriano. La respuesta plantar fue extensora en forma bilateral. También se evocaron reflejos de Trómner y Hoffman en forma bilateral. La exploración cerebelosa resultó normal y no había signos de irritación meníngea.
Laboratorio
• Glucosa 110 mg/dL, BUN 18.1mg/dL urea 38.5 mg/ dL creatinina 0.8 mg/dL.
• Leucocitos 13900, 12% linfocitos, 82.8% polimorfonucleares.
• LCR: Incoloro, células: 2 / mm3 proteínas: 65 mg/dL, glucosa 60 mg/dL.
• Perfil inmunológico normal.
• EGO: leucocituria, levaduras y bacterias muy abundantes.
• VIH negativo.
Gabinete
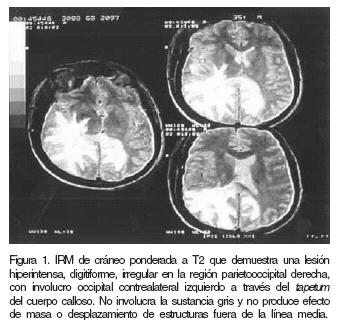
Se realizó estudio de resonancia magnética (IRM) de cráneo con gadolinio que mostró imagen hipointensa en T1 e hiperintensa T2 (Figura.1), siendo esta irregular, digitiforme, respetando la sustancia gris y las fibras arcuatas y abarcando la región parietooccipital derecha , con diseminación occipital izquierda a través del tapetum del cuerpo calloso, sin efecto de masa o desplazamiento de estructuras fuera de la línea media.
Comentario clínico
Con los datos mencionados en la historia clínica y con los hallazgos del examen neurológico se pueden elaborar las siguientes conclusiones sindromáticas:
1. Síndrome piramidal bilateral
Babinski bilateral. Hoffman y Trówner bilateral.
2. Síndrome frontal
Cambios de personalidad, labilidad emocional, fallas de memoria
3. Síndrome parietal–occipital derecho Hemianopsia homónima izquierda, pérdida del reflejo optocinético
4. Síndrome medular transverso completo Paraplejia flácida, arreflexia osteotendinosa de miembros inferiores, nivel sensitivo T7 para todas las modalidades, ausencia de reflejos cutáneos abdominales y cremasteríanos, Babinski bilateral, trastorno esfinteriano.
Diagnóstico anatomotopográfico
1. Las manifestaciones corticales orientan hacia una localización parietal y occipital derecha por la presencia de hemianopsia homónima izquierda y la pérdida del reflejo opto cinético hacia ese lado.
2. Probablemente exista involucro frontal secundario a compromiso de fibras de asociación manifestadas por la presencia de cambios de personalidad, labilidad emocional y fallas de memoria.
3. Las manifestaciones medulares orientan a una lesión medular transversa de tipo mielítba, según el consorcio internacional para el diagnóstico de la misma.5
Diagnóstico integral
El presente caso se trata de un hombre de 35 años de edad, con antecedente de cuadro rinofaríngeo 15 días previos a su padecimiento actual que inicia con manifestaciones neurológicas en dos sitios diferentes del neuroeje, encéfalo y médula, que sustentan el diagnóstico nosológico de encefalomielitis diseminada aguda cuya etiología debe determinarse siempre. Dicha entidad requiere un protocolo de estudio, principalmente diferencial, que incluye procesos postinfecciosos virales, bacterianos, postvacunales, enfermedades primarias o secundarias de la mielina, como esclerosis múltiple, leucoencefalopatia multifocal progresiva, leucodistrofia metacromática, adrenoleucodistrofias, enfermedades inmunológicas como lupus eritematoso sistémico, vasculitis primaria y secundaria del sistema nervioso central, algunas entidades como sarcodosis y encefalitis por virus del síndrome de inmunodeficiencia adquirida.2,3,6 En los casos en los que las manifestaciones clínicas y paraclínicas (laboratorio, inmunología, estudio completo del LCR, etc) no demuestran una etiología específica, es necesario realizar biopsia guiada con cirugía estereotáxica, como sucedió en el caso que nosotros estudiamos.
Hallazgos anatomopatológicos
Se analizaron fragmentos de tejido nervioso, tomados vía biopsia guiada por cirugía estereotáxica, predominantemente de corteza parietal y occipital, con los siguientes hallazgos histológicos de importancia:
1. Se encontraron datos de edema cerebral moderado, así como células de la glia reactivas y aumentadas de volumen, astrocitosis y oligodendrocitos edematizados. (Figura 2).

2. Se observó también infiltrado linfocitario y escasos leucocitos polimorfonucleares, perivasculares,
3. alrededor de dos vasos sanguíneos y probablemente vasculitis. (Figura 3).

Finalmente se apreciaron también cuerpos de inclusión virales intranucleares en neuronas. (Figura 4).

Diagnósticos anatomopatológicos finales
Proceso inflamatorio subagudo de tipo viral con cuerpos de inclusión sugerentes de encefalitis herpética.
Correlación clínico patológica
La encefalomielitis diseminada aguda (EMDA) es una condición desmielinizante aguda diseminada que afecta principalmente el encéfalo y la médula espinal. Generalmente es secundaria a una infección o vacunación. Se caracteriza por lesiones multifocales de la sustancia blanca en neuroimagen. Es mas frecuente en niños y adultos jóvenes; la incidencia de EMDA postinfecciosa posterior a una infección por sarampión es de 1:1000. Las infecciones que generalmente se asocian a EDMA son cuadros virales inespecíficos del aparato respiratorio superior1,3 aunque la gama de patologías que puede producirla incluye procesos bacterianos, virus de la deficiencia adquirida, enfermedades de la mielina, vasculitis y otras enfermedades menos comunes.6
Los agentes etiológicos involucrados más frecuentemente incluyen: sarampión, adenovirus, varbela, hepatitis A y B, rubéola, citomegalovirus, enterovirus, Chlamidia, Epstein–Barr, Legionella, HTLV1, Campilobacter, Herpes tipo 6, Mboplasma pneumoniae, Herpes simplex, Listeria monocytogenes, influenza A y B, Leptospira Interrogans, entre otros.
La presentación clínica de EMDA comienza frecuentemente 1 a 4 semanas después de un cuadro infeccioso o de una inmunización, sin embargo estos antecedentes pueden estar ausentes hasta en un 30% de los casos.
Las principales características de pacientes con EMDA son: edad promedio de presentación a los 33 años y predominio en el sexo femenino. En la EMDA postinfecciosa, los síntomas se inbian cuando ocurre la resolución del cuadro febril y se caracterizan por cefalea, convulsiones y encefalopatía.3 Los signos mas frecuentes en este padecimiento son déficit motor (hemiparesia, paraparesia), déficits sensitivos, alteración de esfínteres, mioclonlas, neuritis óptica, oftalmoplejia, coreoatetosis, mutismo y signos meníngeos. Se pueden presentar psicosis agudas en casos atípicos.2,3
Las manifestaciones encefálicas (encefalitis) ocurren en 90–96% de los casos y las medulares (mielitis) en el 5 al 10%; puede haber polirradiculitis en el 1 al 7% de bs pacientes3 y se han descrito otros síndromes neurobgicos característicos de la EMDA como la mielitis post sarampión, la ataxia cerebelosa post varicela y la encefalopatía post rubéola.
La evolución general de los pacientes en esta enfermedad es la presentación y progresión del cuadro neurológico en días a semanas (hasta un mes) y posteriormente comienza la recuperación, de velocidad y grado variable. Existen casos de progresión más prolongada y de recaídas hasta 2 años después del cuadro inicial, pudiendo o no asociarse a nuevos cuadros infecciosos o post inmunización, por lo que se recomienda no estimular el sistema inmunologico con vacunas hasta seis meses de haber presentado el cuadro inicial.4
El líquido cefalorraquídeo (LCR) es normal en el 30–70% de los casos, pero puede haber pleocitosis y Mocitos menores a 300. Las proteínas pueden estar elevadas y la proteína básica de mielina está aumentada en el 70% de los casos.34 Los hallazgos radiológicos no son patognomónicos. En la fase aguda de la ADEM la TAC es normal hasta en un 40% de bs casos, sin embargo, en nuestro paciente la TAC mostraba una imagen hipodensa de la sustancia blanca. La IRM craneal y de médula espinal es el método de elección para demostrar el compromiso parenquimatoso precoz. Las alteraciones que se han descrito en IRM son imágenes hipointensas en la secuencia T1 e hiperintensas en la secuencia T2 y la administración de medb de contraste produce reforzamiento de algunas lesiones, lo cual refleja ruptura de la barrera hemato–encefálica. En algunos casos este reforzamiento puede ser anular, simulando un tumor o absceso cerebral. Las lesbnes de la sustancia blanca son difusas, asimétricas y habitualmente bilaterales. También se puede afectar la sustancia gris a nivel de bs ganglios básales y tálamo.4,8 En nuestro paciente las lesbnes se confinaron a la regbn temporo–parieto–occipital derecha y occipital izquierda, involucrando las fibras del tapeium del cuerpo calloso.
El electroencefalograma generalmente es anormal, pero inespecífico. Puede mostrar asimetría difusa de alto voltaje en rangos Hiela y delta, ritmo alfa o un patrón alternante. Los potenciales evocados somatosensoriales y visuales pueden alterarse si existe compromiso de la médula espinal o tracto óptico, respectivamente.3,4
El tratamiento más utilizado son los esteroides, debido a su potente efecto antinflamatorio e inmunosupresor. Otras alternativas son el uso de plasmaferesis o de inmunoglobulina intravenosa.3,4
La recuperación completa se obtiene en 50% de los casos. Son factores de mal pronóstico el inicio hiperagudo, las convulsiones y el coma. Un punto interesante en este tipo de patología, es que puede ser el cuadro de presentación inbial de esclerosis múltiple (EM) y se ha publicado que hasta un 35% de los casos desarrollarán una EM,1 siendo los factores predictivos para esta evolución la ausencia de infección asociada, pleocitosis escasa y presencia de bandas oligoclonales en el LCR. Las lesiones en IRM que tienen valor predictivo para el desarrollo de EM son: compromiso peri ventricular, existencia simultánea de lesiones que captan y no captan contraste y lesiones infratentoriales.3 Nuestro paciente presentó un caso típico de encéfalomielitis postinfecciosa aguda, tanto por clínica como por hallazgos de IRM, además de buena recuperación, como se describe en la literatura.
Nuestro caso ejemplifica la presentación clínica típica de una EMDA con cuadro confusional, defecto campimétrico (hemianopsia homónima izquierda) y síndrome medular completo a nivel T7 (mielitis), además del antecedente previo de infección de vías respiratorias superiores.
En nuestro paciente no había el antecedente de inmunización reciente ni de infección por sarampión y el reporte histológico demostró que las lesiones de la IRM y medulares fueron producidas por el virus herpes zoster, como en algunos reportes de la literatura.3,4
Nosotros consideramos que en pacientes con manifestaciones encefálicas y medulares agudas o subagudas el diagnóstico diferencial debe considerar a la EMDA, a la esclerosis múltiple y a enfermedades que afecten simultáneamente la sustancia blanca en varios niveles del sistema nervioso.
Referencias
1. Gag RK. Acute disseminated encephaiomyeiitis. Postgrad Med J 2003;79:11–17. [ Links ]
2. Schwarz S, Mohr A, Knauth M, et al. Acute disseminated encephalomyelitis: A follow–up study of 40 adult patients. Neurology. 2001; 56: 1313–1318. [ Links ]
3. Tenerbaun S, Chamóles N, Firman N. Acute disseminated encephalomyelitis A long–term follow–up study of 84 pediatric patients. Neurology 2002;59:1221–1231. [ Links ]
4. Lucchinetti C. The spectrum of idiopathic inflammatory demyelinanting disease. En: American Academy of Neurology. Syllabi on CD ROM. 2000. [ Links ]
5. Bennetto L, Scolding L. Inflammatory post–infectuos encephalomyelitis. J Neurol Neurosurg Psychiatry 2004; 75(Suppl 1):i22–i28 [ Links ]
6. Douglas K. Harold Y. The Transverse Myelitis consortium working group. Curr Opin Neurol 2002;15:339–47. [ Links ]
7. Stüv O, Scott Z. Diferential diagnosis of inflammatory demyelinating disease. Curr Opin Neurol 2002; 12:395–401. [ Links ]
8. Hynson JL, Kornberg AJ, Coleman LT, et al. Clinical and neuroradiologic features of acute disseminated encephaiomyelitis in children. Neurology 2001; 56:1308–1312. [ Links ]

