










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta médica de México
versión On-line ISSN 2696-1288versión impresa ISSN 0016-3813
Gac. Méd. Méx vol.140 no.1 Ciudad de México ene./feb. 2004
Artículos de revisión
Uso de la vasopresina en el estado de choque
Vasopressin Use in Shock
Raúl Carrillo–Esper,* Jorge A. González–Salazar,** Benjamín Calvo–Carrillo**
* Profesor titular de postgrado de Terapia Intensiva UNAM, Jefe de la Unidad de Terapia Intensiva. Hospital Central Sur de Alta Especialidad PEMEX.
** Residentes de la Unidad de Terapia Intensiva.
Correspondencia:
Hospital Central Sur de Alta Especialidad PEMEX
Picacho. Periférico Sur No. 4091.
México, D.F.
Tel y Fax: 56 45 16 84 Ext: 51155.
Recepción versión modificada 04 de julio del 2002;
Aceptación 17 de julio del 2002
Resumen
La arginina–vasopresina (VP) también conocida como hormona antidiurética es esencial para mantener el equilibrio hídrico. Su síntesis y liberación depende de la interacción de estímulos osmóticos, hipovolémicos, hormonales y no osmóticos. Se ha demostrado que en estados de choque es fundamental para mantener la homeostasis cardiovascular a través de la regulación del tono vasomotor, el cual determina las resistencias vasculares sistémicas y la presión arterial media, a través de los receptores VI. El estado de choque con vasodilatación refractaria que se presenta en sepsis, respuesta inflamatoria sistémica, hipovolemia, paro cardiaco, politraumatismo, etc... se caracteriza por una fase inicial en la que hay liberación y aumento en los niveles séricos de VP, ésta es seguida por una segunda fase en la que se presentan niveles inapropiadamente bajos de la hormona y éstos se asocian con refractariedad al manejo con volumen, inotrópicos y vasopresores. Se ha demostrado, en estudios experimentales y clínicos, que en esta condición el tratamiento con vasopresina exógena incrementa la resistencia vascular sistémica, la presión de perfusión y el aporte de oxígeno a los tejidos periféricos lo cual hace posible la disminución y suspensión de los vasopresores e incrementa la supervivencia.
Palabras clave: Vasopresina, choque refractario, vasodilatación refractaria, paro cardiaco
Summary
Arginine–vasopresin (VP), also known as the antidiuretic hormone, is essential for water homeostasis. Its synthesis and liberation depends on regulation of osmotic, hypovolemic, hormonal, and nonosmotic stimuli. It has been demonstrated that it is key for maintenance of cardiovascular homeostasis through vasomotor regulation, the determinant of systemic vascular resistance and mean arterial pressure, a process acting through VI receptors. Shock state with refractary vasodilation seen in sepsis, systemic inflamatory response, hypovolemia, cardiac arrest, polytrauma, etc., is characterized by an initial phase of liberation and increased levels of VP followed by a second phase caracterized by inappropirately low levels of this hormone that are associated with refractariness to management with volume, inotropics, and vasopressors. It has been demonstrated in clinical and experimental studies that exogenous VP treatment under this condition increases systemic vascular resistance, perfusion pressure, and oxygen supply to peripheral tissues, which makes it possible to decrease and to suspend vasopressors and also to increase survival.
Key words: Vasopressin, refractary shock, refractary vasodilatation states, cardiac arrest
La arginina–vasopresina (VP) también conocida como hormona antidiurética es esencial para mantener el equilibrio hídrico y la estabilidad cardiovascular. En los últimos años su aplicación clínica se ha centrado en el manejo de la diabetes insípida y de la hemorragia por várices esofágicas.
En 1971 Errington describió la cinética de la VP en el choque hemorrágico, a partir de entonces aparecieron en la literatura múltiples estudios relacionados con los cambios y efectos de la VP en el estado de choque. En 1997 Landry y col. propusieron su uso en el choque séptico refractario y en otros estados de choque asociados a vasodilatación como el que se presenta después de la colocación de un puente aortocoronario. Por los efectos vasculares de la VP y el incremento asociado en la perfusión coronaria, Wenzel y col. la propusieron como medicamento promisorio en la reanimación cardiopulmonar.1–3
El objetivo del presente trabajo es dar a conocer a la comunidad médica las aplicaciones terapéuticas de la VP en el paciente críticamente enfermo.
Fisiología
Síntesis y liberación
La VP es un nonapéptido con un puente disulfuro entre dos cisternas. El gen que codifica para la síntesis de VP está constituido por tres exones y en el humano se localiza en el cromosoma 20.4
Se sintetiza en las neuronas magnocelulares localizadas en los núcleos supraóptico y para ventricular del hipotálamo como pre–prohormona la cual está constituida por el nonapéptido arginina vasopresina (AVP), neurofisina II, aminoácidos básicos, copeptina y un tripéptido de unión. Es transportada del citoplasma al aparato de Golgi donde se almacena en granulos neurosecretorios. Ahí son separados los diferentes componentes y transportados vía axonal a la hipófisis posterior, en donde se almacenan. Únicamente de 10–20% de la hormona es liberada rápidamente, después se libera en pulsos lentos y continuos. El proceso completo de síntesis, transporte y almacenaje se lleva a cabo en una a dos horas.5,6
La liberación de la VP es secundaria a estímulos osmóticos, hipovolémicos, hormonales y no osmóticos.7–10
Regulación osmótica
La hiperosmolaridad es uno de los estímulos más potentes para la liberación de VP. Es controlada por la activación de osmorreceptores periféricos y centrales. Los osmorreceptores periféricos se encuentran localizados en la vena porta y responden de manera rápida a los cambios osmolares inducidos por los alimentos y líquidos ingeridos. Los osmorreceptores centrales se encuentran localizados en regiones cerebrales excluidas de la barrera hematoencefálica detectan cambios de osmolaridad en margenes muy estrechos. La activación de receptores por cambios osmolares induce despolarización de las neuronas magnocelulares de los núcleos supraóptico y paraventricular iniciando el proceso de transporte y liberación de VP.
Regulación hipovolémica
La depleción de volumen intravascular y la hipotensión, estimulan la liberación de VP a través de la activación de receptores de estiramiento que se encuentran localizados en aurícula izquierda, ventrículos, arco aórtico y seno carotídeo. Los dos primeros registran los cambios de volumen y los segundos registran los de presión. La activación de estos receptores además de la liberación de VP, inicia los mecanismos adrenérgicos, liberación de renina y modulación del péptido natriurético auricular con la finalidad de mantener estabilidad hemodinámica.11–13
Regulación hormonal
Hay una serie de hormonas y mediadores que se liberan en el estado de choque y que estimulan o inhiben directamente la liberación de VP. Los factores liberadores son: acetilcolina (vía receptores nicotínicos), histamina, nicotina, dopamina, prostaglandinas, angiotensina 11. La hipoxemia y la hipercapnia a través de la estimulación de quimiorreceptores localizados en el cuerpo carotídeo son potentes liberadores de VP.
Los inhibidores descritos hasta el momento son: opioides, ácido gamma–amininobutírico, péptido natriurético auricular.
El óxido nítrico (ON) es mediador de vasodilatación e hipotensión en choque séptico, además tiene la capacidad de inhibir la liberación de VP vía AMPc.13
La norepinefrina (NE) tiene acción bifásica porque a través de receptores alfa 1 o alfa 2 inhibe o estimula la secreción de VP.14,15
Regulación no osmótica
Dentro de ésta se incluyen al dolor, la náusea, la acidosis y el estrés emocional. La náusea puede incrementar los niveles de VP, de 20 a 500 veces, y contribuye a la elevación de ésta durante reacciones vasovagales, vértigo de movimiento, hiperemesis del embarazo, quimioterapia y cetoacidosis.16
Metabolismo
Los niveles séricos normales de VP en estado de hidratación adecuada son < 4 pg/mL, mientras que la deprivación de agua y el incremento de la osmolaridad plasmática se asocian a niveles de 10 pg/mL. La concentración máxima de orina se logra con niveles de VP de 20 pg/mL. Su vida media es de diez a treinta y cinco minutos y es metabolizada por vasopresinasas en hígado y riñon.17
Receptores de vasopresina
Los receptores de vasopresina pertenecen a la superfamilia de receptores de proteína G con siete dominios transmembrana. Se han descrito cuatro subtipos de receptores cuya localización, densidad y distribución determinan los diferentes efectos fisiológicos. (Cuadro I).
Los receptores V1 se localizan en el endotelio vascular y median la vasoconstricción por activación de fosfolipasa C y liberación de calcio por la vía de fosfoinositol. Los receptores V2 se localizan en túbulos colectores renales y células endoteliales y median la retención de agua y el efecto antidiurético de la VP a través de la activación de adenilato ciclasa y el incremento de AMP cíclico. Los receptores V3 tienen efectos centrales e incrementan los niveles de ACTH a través de la activación de diferentes proteínas G. Los receptores de oxitocina (OTR) se localizan en útero, mama, células endoteliales de vena umbilical, aorta y vena pulmonar. Su activación induce la contracción uterina y participa en la respuesta vasodilatadora dependiente de calcio, mediante la estimulación de óxido nítrico (ON).18,19
Efectos sistémicos
Las acciones fisiológicas de la VP son las siguientes:
a Regula el metabolismo del agua mediante el aumento en la permeabilidad de los túbulos colectores a través de receptores V2, esto a su vez regula la retención hídrica y su efecto antidiurético.
b Regula el tono vasomotor y de esta manera interviene en la estabilidad hemodinámica.
c Favorece la liberación de ACTH y cortisol.
d A través de activación de receptores V2 (agonista sintético 1 –desamino–8–Darginina vasopresina) causa agregación plaquetaria y liberación del factor de Von–Willebrand.
e A nivel cerebral actúa como neurotransmisor involucrado en: ritmos circadianos, ingesta de agua, regulación cardiovascular, termorregulación y nocicepción.20
Vasopresina en el estado de choque
El efecto antidiurético y la conservación de agua constituye la función más notable de la VP, pero también ésta participa en la homeostasis cardiovascular a través de vasoconstricción, mantenimiento de resistencias vasculares sistémicas y presión arterial. Su efecto antidiurético se observa en márgenes de 1 –7 pg/mL, y su efecto vasoconstrictor requiere de niveles de 10–200 pg/ mL21,22
La VP es un potente vasoconstrictor en piel, músculo esquelético, grasa, páncreas y tiroides, y su efecto es menor en la circulación mesentérica, coronaria y cerebral, lo cual puede estar en relación con su interacción con ON.
En condiciones fisiológicas la VP juega un papel menor en la regulación de la presión arterial, pero durante el choque hemorrágico o séptico es fundamental para mantener presión arterial. Lo anterior se ha demostrado en modelos experimentales de choque endotóxico en los cuales el pretratamiento con antagonistas específicos de VP amplifica el efecto vasodilatador e hipotensor de la endotoxina, asimismo, el tratamiento con VP exógena incrementa la presión arterial y disminuye la mortalidad. Por otra parte, animales con diabetes insípida presentan mala respuesta a la hipotensión y a los estados de choque con elevada mortalidad.23–28
En el estado de choque la VP tiene una respuesta bifásica. En la primera fase se presenta incremento en su liberación con niveles séricos que varían entre 100 a 1800 pg/mL, mientras que en la segunda fase los niveles séricos descienden progresivamente hasta 1–12 pg/mL, lo cual se asocia a choque, vasodilatación refractaria e hipoperfusión coronaria. Se ha demostrado en múltiples estudios que esto se relaciona con mal pronóstico y falta de respuesta al manejo con líquidos, inotrópicosy vasopresores.29–34
Este fenómeno se ha descrito en choque séptico, hemorrágico, en donadores de órganos con grave inestabilidad hemodinámica y en estados de vasodilatación posteriores a la colocación de un puente aorto–coronario de un dispositivo de asistencia mecánica–ventricular. Esto sugiere que en estados de choque asociados a vasodilatación refractaria existe depleción relativa de VP o lo que también se ha denominado, niveles plasmáticos inapropiadamente bajos, en los cuales a pesar de que se preserva la acción antidiurética se pierde el efecto modulador del tono vascular.34–36
La disminución en los niveles séricos de VP que se presentan en los estados de choque ya descritos es de etiología multifactorial:
a Depleción de VP en hipófisis posterior secundaria a intensa estimulación osmótica y barorreceptora.
b Insuficiencia autonómica con bloqueo de barorreceptores periféricos, lo cual se asocia a interferencia con la señal disparadora a nivel central,
c Los niveles séricos elevados de norepinefrina, ya sea como parte de la respuesta metabólica al estado de choque o secundarios a su uso terapéutico, condicionan inhibición en la liberación de VP a nivel central a través de los receptores alfa 1.
d El incremento en la síntesis de ON por el endotelio vascular de la hipófisis posterior con la subsecuente inhibición en la liberación de VP, interfiere con su efecto vasopresor periférico. Dicho proceso se amplifica por la producción exagerada de ON a nivel sistémico y por el efecto sinérgico entre la hipoxia y los niveles bajos de VP sobre la apertura de los canales de K dependientes de ATP. (Figura 1).
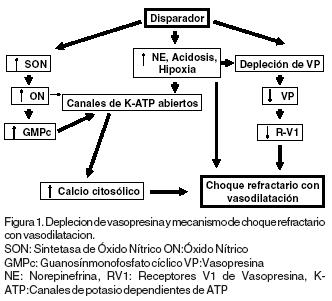
Uso terapéutico de la vasopresina en el estado de choque
La corrección de los niveles inapropiadamente bajos de VP mediante la aplicación exógena de esta hormona se ha postulado como alternativa terapéutica en enfermos con choque y vasodilatación refractarias al empleo de volumen y vasopresores.
La VP ejerce su acción vasopresora en choque hipovolémico ó séptico en fase de vasodilatación aun en casos de resistencia a norepinefrina, angiotensina II y endotelina.37,38
Los mecanismos involucrados en el efecto hemodinámico de la VP en el estado de choque son:39–46
a Las concentraciones inapropiadamente bajas de VP favorecen una mayor disponibilidad de receptores V1, de tal manera que aun a dosis bajas, la VP los ocupa y ejerce su acción vasoconstrictora,
b La VP potencia el efecto vasoconstrictor de la NE.
c La VP inhibe los canales de potasio (K) dependientes de ATP en el músculo liso vascular y por eso incrementa la disponibilidad de calcio celular necesario para la contracción del músculo liso,
d La VP bloquea al GMPc que es el segundo mensajero del ON, bloqueando así su acción vasodilatadora,
e La VP bloquea la síntesis de la sintetasa inducible de ON mediada por lipopolisacárido y la liberación de péptido natriurético auricular.
Ensayos clínicos
El estado de estado de choque en fase de vasodilatación, refractaria a volumen, inotrópicos y vasopresores se caracteriza por un mal pronóstico. El estudio de Landry demostró que la vasopresina es de utilidad en esta fase del estado de choque.
A partir de entonces y hasta 2001 se han publicado doce estudios clínicos que incluyen a 212 enfermos en estado de choque en fase de vasodilatación refractaria, de diferente etiología (sepsis, respuesta inflamatoria sistémica, postpuente aortocoronario, traumatismo, hipovolemia, secundario a inhibidores de la fosfodiesterasa y choque en pacientes pediátricos) que respondieron a la infusión de VP a dosis de 0.01 –0.04 Ul/min. Los niveles séricos de VP oscilaban entre 2.9 a 20 pg/mL antes del tratamiento y una vez iniciada la infusión alcanzaron valores de 50 a 300 pg/mL.47–55 Estos estudios demostraron que el empleo de VP incrementa de manera significativa la resistencia vascular sistémica y la presión de perfusión, y mejora el aporte de oxígeno a los tejidos periféricos, lo cual se asoció con una disminución significativa en la mortalidad en comparación con grupos control que no recibían VP. Se asocia con incremento en la diuresis, el cual está en relación con una mejor perfusión renal y del lecho esplácnico, con el efecto natriurético y con la regulación de la liberación de otros mediadores como: péptido natriurético auricular, renina, angiotensina II y aldosterona.56
La infusión de vasopresina debe mantenerse hasta lograr estabilidad hemodinámica y disminución en la dosis de las aminas vasoactivas. Algunos reportes mencionan que esto se logra en periodos de tiempo que van de 2 a 284 h. La vasopresina, debe suspenderse progresivamente, pues de lo contrario se puede volver a presentar el deterioro hemodinámico.56,57
Las infusiones de vasopresina se asocian a vasoconstricción e isquemia renal, esplácnica y coronaria. Esta complicación se ha descrito en enfermos con hemorragia por várices esofágicas en quienes se utilizaron dosis elevadas de VP, pero no se ha reportado en los casos en que se administró en dosis bajas para el manejo del estado de choque con vasodilatación refractaria; sin embargo, se recomienda que durante su administración se vigile estrechamente la perfusión coronaria y esplácnica mediante monitoreo de la onda T y el segmento ST, enzimas cardiacas, pruebas de funcionamiento hepático, determinación de azoatos, depuración de creatinina y datos clínicos de isquemia intestinal.58
Vasopresina en paro cardiaco
El paro cardiaco y la actividad eléctrica sin pulsos se caracterizan por pérdida del tono vascular periférico y estado de choque con vasodilatación, lo cual trae como consecuencia una mala distribución del flujo sanguíneo e hipoperfusión coronaria. La epinefrina (E), por su efecto vasoconstrictor, es considerada el medicamento de elección en los algoritmos de reanimación, porque incrementa la resistencia vascular sistémica y la perfusión coronaria.
En el paro cardiaco la VP presenta la misma dinámica que en los estados de choque comentados previamente, pueden presentarse niveles séricos inapropiadamente bajos para los requerimentos hemodinámicos. Por este motivo desde 1997 se postuló que la VP podría ser útil para las maniobras de reanimación en paro cardiaco.
La utilidad de la VP durante el paro cardiaco se ha demostrado tanto en estudios animales como humanos, en éstos se ha observado que incrementa el flujo sanguíneo miocárdico y cerebral, y mejora la sobrevida.59–62
En estudios comparativos entre VP y E, se demostró que aquellos pacientes que son manejados con VP tienen mejor respuesta a la reanimación cardiopulmonar y una mayor supervivencia. Por lo anterior, en la actualidad la American Heart Association la recomienda como alternativa a la E en fibrilación ventricular refractaria y como medicamento de rescate en aquellos enfermos en paro cardiaco a pesar del uso de dosis altas de E. Se requiere un mayor número de estudios; sin embargo, la VP parece un medicamento promisorio que puede llegar a ser de primera elección en las maniobras de reanimación en paro cardiaco.63–68
Conclusiones
El choque refractario con vasodilatación es secundario a diferentes entidades y cursa con niveles inapropiadamente bajos de VP. La corrección de éstos con VP exógena, a dosis de 0.01–0.04 Ul/min incrementa la resistencia vascular sistémica y la presión de perfusión, lo cual mejora el aporte de oxígeno y la superviviencia de los enfermos.
Referencias
1. Errington ML, Rocha M, Silva E Jr. The secretion and clearance of vasopressin during the development of irreversible haemorrhagic shock. Proc Physiol Soc 1971; 23:43P–45P. [ Links ]
2. Landry DW, Levin HR, Gallant EM, et al. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation 1997; 95:1122–1125. [ Links ]
3. Wenzel V, Prengel AW, Maier C, et al. Vasopressin improves vital organ blood flow after prolonged cardiac arrest with postcountershock pulseless electrical activity in pigs. Crit Care Med 1999; 27:486–492. [ Links ]
4. Ridell DC, Mallonee R, Philips JA, et al. Chromosomal assignment of human sequences encoding arginine vasopressin–neurophysin II and growth hormone releasing factor. Somat Cell Mol Genet 1985; 11:189–195. [ Links ]
5. Robertson GL. The regulation of vasopressin function in health and disease. Prog Horm Res 197; 33:333–385. [ Links ]
6. Physiological pathways regulating the activity of magnocellular neurosecretory cells. Prog Neurobiol 1999; 57:625–655. [ Links ]
7. Sklar AH, Schrier R. Central nervous system mediators of vasopressin release. Physiol Rev 1983; 63:1243–1280. [ Links ]
8. Bourque CW, Oliet SH, Richard D. Osmoreceptors, osmoreception and osmoregulation. Front Neuroendocrinol 1994;15:231 –274. [ Links ]
9. Schrier RW, Berl T, Anderson RJ. Osmotic and nonosmotic control of vasopressin release. Am J Physiol 1979; 236:F321–F332. [ Links ]
10. Robertson GL, Shelton RL, Athar S. The osmoregulation of vasopressin. Kidney Int 1976;10:25–37. [ Links ]
11. Thrasher TN. Baroreceptor regulaton of vasopressin and renin secretion: low–pressure versus high–pressure receptors. Front Neuroendocrinol 1994: 15:157–196. [ Links ]
12. Wang BC, flora–Ginter G, Leadley RJ Jr, et al. Ventricular receptors stimulate vasopressin release during hemorrhage. Am J Physiol 1988; 254:R204–R211. [ Links ]
13. Reid IA. Role of nitric oxide in the regulation of renin and vasopressin secretion. Front Neuroendocrinol 1994; 15:351–383. [ Links ]
14. Day TA, Randle JC, Renaud LP. Opposing alpha–and beta–adrenergic mechanisms mediate dose–dependent actions of norepinephrine on supraoptic vasopressin neurones in vivo. Brain Res 1985; 358:171–179. [ Links ]
15. Randle JC, Bourque CW, Renaud LP. Alphai –adrenergic receptor activation depolarizes rat supraoptic neurosecretory neurons in vitro. Am J Physiol 1986: 251 :R569–R574. [ Links ]
16. Kovacs L, Robertson GL. Syndrome of inappropriate antiduresis. Endocrinol Metab Clin North Am 1992; 21:859–875. [ Links ]
17. Share L, Kimura T, Matsui K, et al. Metabolism of vasopressin. Fed Proc 1985: 44:59–61. [ Links ]
18. Barberis C, Mouillac B, Durroux T. Structural bases of vasopressin/oxytocin receptor function. J Endocrinol 1998; 156:223–229. [ Links ]
19. Bichet D. Vasopressin receptors in health and disease. Kidney 1996; 49:1706–1711. [ Links ]
20. Riphagen CL, Pttman QJ. Arginine vasopressin as a central neurotransmitter. Fed Proc 1986; 45:2318–2322. [ Links ]
21. Czaczkes JW. Physiologic studies of antidiuretic hormone by its direct measurement in human plasma. J Clin Invest 1964; 43:1625–1640. [ Links ]
22. Morton JJ, Padfield PL, Forsling ML. A radioimmunoassay for plasma arginine–vasopressin in man and dog: application to physiological and pathological states. J Endocrinol 1975;65:411–424. [ Links ]
23. Arnauld E, Czernichow P, Fumoux F, et al. The effects of hypotension and hypovolaemia on the liberation of vasopressin during hemorrhage in the unanaesthetized monkey (Macaca mulatta). Pflugers Arch Eur J Physiol 1977; 371:193–200. [ Links ]
24. Cowley AW Jr, Switzer SJ, Guinn MM. Evidence and quantification of the vasopressin arterial pressure control system in the dog. Circ Res 1980; 46:58–67. [ Links ]
25. Bracken DJ, Schaefer CF, Wilson MF. The role of vasopressin in the maintenance of cardiovascular function during early endotoxin shock. Adv Shock Res 1983; 9:147–156. [ Links ]
26. Zerbe RL, henry DP, Robertson GL. Vasopressin response to orthostatic hypotension: etiologic and clinical implications. Am J Med 1983; 74:265–271. [ Links ]
27. Schwartz J, Reid IA. Role of vasopressin in blood pressure regulation in conscious water–deprived dogs. Am J Physiol 1983; 244:R74–R77. [ Links ]
28. Mohnring J, Glanzer K, Maciel JA Jr, et al. Greatly enhanced pressor response to antidiuretic hormone in patients with impaired cardiovascular 51. reflexes due to idiopathic orthostatic hypotension. J Cardiovasc Pharmocol 1980; 2:367–376. [ Links ]
29. Wilson MF, Brackett DJ, Tompkins P, Benjamin B, Archer LT, Hinshaw LB. Elevated plasma vasopressin concentrations during endotoxin and E. Coil shock. Adv Shock Res 1981; 6:15–26. [ Links ]
30. Baker CH, Sutton ET, Zhou Z, Dietz JR. Microvascular vasopressin effects during endotoxin shock in the rat. Circ Shock 1990; 30:81–95. [ Links ]
31. Brackett DJ, Schaefer CF, Wilson MF. The role of vasopressin in the maintenance of cardiovascular function during early endotoxin shock. Adv Shock Res 1983; 9:147–56. [ Links ]
32. Wilson MF, Brackett DJ, Hinshaw LB, et al. Vasopressin release during sepsis and septic shock in babonsand dogs. Surg Gynecol Obstet 1981; 153:869–872. [ Links ]
32. Errington ML, Rocha e Silva M Jr. The secretion and clearance of vasopressin during the development of irreversible hemorrhagic shock. J Physiol (Lond) 1971; 217:43P–45P. [ Links ]
33. Brackett DJ, Schaefer CF, Tompkins P, et al. Evaluation of cardiac output, total peripheral vascular resistance, and plasma concentrations of vasopressin in the conscious, unrestrained rat during endotoxemia. Circ Shock 1985; 17:273–284. [ Links ]
34. Landry DW, Levin HR, Gallant EM, et al. Vasopressin pressor hypersensitivity in vasodilatory septic shock. Crit Care Med 1997; 25:1279– 1282. [ Links ]
35. Argenziano M, Choudhri AF, Oz MC, et al. Prospective randomized trial of arginine vasopressin in the treatment of vasodilatory shock after left ventricular assist device placement. Circulation 1997; 96:11–286–290. [ Links ]
36. Argenziano M, Chen JM, Choudhri AF, et al. Management of vasodilatory . shock after cardiac surgery: identification of predisposing factors and use of a novel pressor agent. Thorac Cardiovasc Surg 1998; 116:973–980. [ Links ]
37. Brackett DJ, Schaefer CF, Wilson MF. The role of vasopressin in the maintenance of cardiovascular function during early endotoxin shock. Adv Shock Res 1983; 9:147–156. [ Links ]
38. Rurak DW. Plasma vasopressin levels during hemorrhage in mature and immature fetal sheep. J Dev Physiol 1979; 1:91–101. [ Links ]
39. Abid O, Akca S, Haji–Michael P, et al. Strong vasopressor support may be futile in the intensive care unit patient with multiple organ failure. Crit Care Med 2000;28:947–949. [ Links ]
40. Matsuoka T, Wisner D. Hemodynamic and metabolic effects of vasopressin blockade in endotoxin shock. Surgery 1997; 121:162–173. [ Links ]
41. Bartelstone HJ, Nasmyth PA. Vasopressin potentiation of cathecholamine actions in dog, rat, cat, and rat aortic strip. Am J Physiol 1965; 208:754– 762. [ Links ]
42. Medina P, Noguera I, Aldasoro M, et al. Enhancement by vasopressin of adrenergic responses in human mesenteric arteries. Am J Physiol 1997; 272:H1087–H1093. [ Links ]
43. Wakatsuki t, Nakaya TY, Inoue I. Vasopressin modulates K–channel activities of cultured smooth muscle cells from porcine coronary artery. Am J Physiol 1992; 263:H491–H496. [ Links ]
44. Umino T, Kusano E, Muto S, et al. AVP inhibits LPS– and IL–1 B–stimulated NO and cGMP via V1 receptor in cultured rat mesangial cells. Am J Physiol 1999; 276:F433–F441. [ Links ]
45. Nambi P, Whitman M, Gessner G, Ajyar N, Crooke ST. Vasopressin–mediated inhibition of atriai natriuretic factor–stimulated cGMP accumulation in an established smooth muscle cell line. Proc Nati Acad Sci USA 1986; 83:8492–5. [ Links ]
46. Lindberg JS, Copley JB, Melton K, et al. Lysine vasopressin in the treatment of refractory hemodialysis–induced hypotension. Am J Nephrol 1990; 10:269–275. [ Links ]
47. Overand PT, Teply JF. Vasopressin for the treatment of refractory hypotension after cardiopulmonary bypass. Anesth Analg 1998; 86:1207– 1209. [ Links ]
48. Gold JA, Cullinane S, Chen J. Vasopressin as an alternativeto norepinephrine in the treatment of milrinone–induced hypotension. Crit Care Med 2000; 28:249–252. [ Links ]
49. Morales D, Madigan U, Cullinane S, et al. Reversal by vasopressin of intractable hypotension in the late phase of hemorrhagic shock. Circulation 1999;100:226–9. [ Links ]
50. Chen JM, Cullinane S, Spanier TB, et al. Vasopressin deficiency and pressor hypersensitivity in hemodynamically unstable organ donors. Circulation 1999; 100:Suppl II:II–244–11–246. [ Links ]
51. Lindner KH, Prengel AW, Brinkmann A, Strohmenger HU, Lindner IM, Lurie KG. Vasopressin administration in refractory cardiac arrest. Ann Intern Med 1996; 124:1061–4. [ Links ]
52. Morales DL, Gregg D, Helman DN, et al. Arginine vasopressin in the treatment of 50 patients with postcardiotomy vasodilatory shock. Ann Thorac Surg 2000; 69:102–106. [ Links ]
53. Chen JM, Cullinane S, Spanier TB, et al. Vasopressin deficiency and pressor hypersensitivity in hemodynamically unstable organ donors. Circulation 1999; 100:11244–11246. [ Links ]
54. Gold J, Cullinane S, Chen J, et al. Vasopressinm in the treatment of milrinone–induced by hypotension in severe heart failure. Am J Cardiol 2000; 85:506–588, A11. [ Links ]
55. Rosenweig EB, Stare TJ, Chen JM, et al. Intraenous arginine–vasopressin in children with vasodilatory shock after cardiac surgery. Circulation 1999; 100:111 82–111 86. [ Links ]
56. Tsuneyoshi I, Yamada H, Kakihana Y, et al. Hemodynamic and metabolic effects of low–dose vasopressin infusions in vasodilatory septic shock. Crit Care Med 2001; 29. [ Links ]
57. Kurtzman NA, Rogers PW, Boonjarern S, et al. Effect of infusion of phamacologic amounts of vasopressin on renal electrolyte excretion. Am J Physiol 1975; 228:890–894. [ Links ]
58. Shelly MP, Greatorex R, Calne RY, et al. The physiological effects of vasopressin when used to control intra–abdominal bleeding. Intensive Care Med 1988; 14:526–531. [ Links ]
59. Voelckel WG, Lindner K, Wenzel V, et al. Effects of vasopressin and epinephrine on splanchnic blood flow and renal function during and after cardiopulmonary resuscitation in pigs. Crit Care Med 2000; 28: [ Links ]
60. Lindner KH, Prengel AW, Pfenninger EG, et al. Vasopressin improves vital organ blood flow during closed–chest CPR in pigs. Circulation 1995; 91:215– 221. [ Links ]
61. Prengel AW, Lindner KH, Keller A. Cerebral oxygenation during cardiopulmonary resuscitation with epinephrine and vasopressin in pigs. Stroke 1996; 27:1241–1248. [ Links ]
62. Lindner KH, Prengel AW, Brinkmann A, et al. Vasopressin administartion in refractory cardiac arrest. Ann Intern Med 1996; 124:1061–1064. [ Links ]
63. Cain BS, Cain JS. Vasopressin in advanced cardiac life support? Crit Care Med 2001; 29. [ Links ]
64. Morris DC, Grzybowski M, Martin GB, et al. Vasopressin can increase coronary perfusion pressure during hyuman cardiopulmonary resuscitation. Acad Emerg Med 1997; 4:878–883. [ Links ]
65. Wenzel V, Krismer AC, Miller EA, et al. Repeated administation of vasopressin but not epinephrine maintains coronary perfusión pressure after early and late administration during prolonged cardiopulmonary resuscitation in pigs. Circulation 1999; 99:1379–1384. [ Links ]
66. Wenzel V, Augenstein S, Voelckel W, et al. Intraosseous vasopressin improves coronary perfusion pressure rapidly during cardiopulmonary resuscitation in pigs. Crit Care Med 1999; 27:1565–1569. [ Links ]
67. Lindner KH, Strohmenger HU, Prengel AW, et al. Rrandomised comparison of epinephrine and vasopressin in patients with out–of–hospital ventricular fibrillation. Lancet 1997; 349:535–537. [ Links ]
68. Voelckel WG, Wenzel V, Bonatti J, et al. Effects of vasopressin and epinephrine on splanchnic blood flow and renal function during and after cardiopulmonary resuscitation in pigs. Crit Care Med 2000; 28:1083– 1088. [ Links ]

