











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink

INTRODUCCIÓN
El estudio y conocimiento de la diversidad genética, el cual detecta las variaciones heredables que pudieran ocurrir en cada organismo, entre los individuos de alguna población y entre poblaciones de una misma especie, cobra relevancia desde hace varias décadas por la diversidad de usos y trabajos que presenta. Para lograr implementar ese tipo de propuestas se requiere de una interrelación entre varias áreas, disciplinas o ciencias. Así, la genética y la evolución se agrupan para entender, describir y dar las pautas en trabajos de biodiversidad, para que sea estudiada o entendida y pueda ser protegida y conservada (Rimieri, 2017).
Alrededor del mundo se están perdiendo diversas especies (Scheffers y col., 2012; Raven, 2020) de flora y fauna silvestre debido a que el ser humano modifica los ambientes a través de la pérdida del hábitat, la contaminación ambiental (presencia de componentes nocivos en el medio ambiente, que pueden ser químicos, físicos o biológicos, ya sea en un entorno natural o artificial), la sobreexplotación, la introducción de especies invasoras, el cambio climático, entre otros (Koslowski y col., 2020). Además del empleo de algunos métodos convencionales (como quemas, generación de dióxido de carbono por industrias, incendios o combustión de automóviles, uso de plásticos, y otros) que aceleraron las alteraciones del ambiente y tal vez la pérdida de alguna especie, por lo que es necesario desarrollar metodologías más precisas que en algunos casos logren distinguir dos especímenes o especies entre sí, para finalmente detectarlos, diferenciarlos y contabilizarlos (Yildiz y col., 2019).
Aunque aún no existe la información completa de la magnitud de dicha pérdida de la biodiversidad, hay un acuerdo internacional llamado Plan Estratégico para la Biodiversidad 20112020, que marca los procedimientos para tratar de detener y revertir la pérdida de la biodiversidad de acuerdo al Programa de Naciones Unidas para el Medio Ambiente (UNEP, por sus siglas en inglés: United Nations Environment Programme) (UNEP, 2011). Estas acciones dependen en gran medida del monitoreo biológico, que permite obtener datos de la especie, lo que se logra normalmente visualizándolas y cuantificándolas individuo por individuo basándose en diversos caracteres morfológicos. El dilema se presenta cuando las especies son similares o idénticas morfológica o anatómicamente (Thomsen y Willerslev, 2015; Deiner y col., 2017). Los errores que se presentan son muy comunes en la clasificación de algunos taxones debido a que se requiere personal altamente entrenado, por lo que se implementan otras técnicas para su identificación y futuro uso de la información (Bortolus, 2008; Hernández-González y col., 2016), como la caracterización molecular.
El campo de la biología molecular cobró relevancia desde el año 2000, dado que entre sus aplicaciones está el estudio de los genomas de organismos, desde un microorganismo hasta organismos superiores, como los humanos. Para entender cómo el estudio genómico de los ácidos nucleicos es utilizado en biodiversidad es necesario señalar que una molécula clave para esos estudios es el ácido desoxirribonucleico (DNA, por sus siglas en inglés: deoxyribonucleic acid), la cual está compuesta por átomos en forma de doble hélice y tiene cuatro bases nitrogenadas llamados nucleótidos: adenina, guanina, citosina y timina. El DNA se encarga de trasmitir la información genética (Vázquez-Ramos, 2016; Koch y col., 2020) y es útil para identificar individuos al comparar secuencias de origen distinto o no.
Con los estudios moleculares del DNA se puede complementar la morfología o la anatomía de especímenes o seres humanos, respectivamente, con lo se obtiene una mayor precisión de la identificación de sujetos (Pizarro, 2003), o mediante el uso de ácido ribonucleico (RNA, por sus siglas en inglés: ribonucleic acid), con el mismo principio de detección del DNA. Estos estudios requieren de cantidades pequeñas de muestra (micro o nanogramos) de ácido nucleico (DNA o RNA) usada para su análisis o amplificación; el protocolo es rápido (menos de 3 h) y está basado en la reacción en cadena de la polimerasa (PCR, por sus siglas en inglés: polymerase chain reaction) (Zhu y col., 2020), o de un par de días para otras técnicas más elaboradas, comparadas con los cultivos de bacterias u hongos (de 1 d a 4 d o más días para hongos de lento crecimiento) (Austin, 2017); versátil (múltiple y/o diferentes tipos de muestras residuales) y, en algunos casos, barato (cuando se consideran pruebas masivas) (Deiner y col., 2017). Asimismo, es utilizada es la secuenciación (Mani, 2020), la cual cuenta con variantes, desde secuenciación química, enzimática, de electroforesis capilar, como la secuenciación tradicional, que por muchos años fue la más común, llamada secuenciación de Sanger, enzimática, capilar o de primera generación (Sanger y col., 1977; van-Dijk y col., 2018), que consume mucho tiempo, y se puede analizar especímen por especimen para obtener una secuencia a la vez. Ésta se basa en la terminación de la amplificación o extención de cadenas de ácidos nucleicos, lo cual produce cadenas de diferentes tamaños que se pueden agrupar por el nucleótido común en donde se terminó la reacción enzimática. Los avances en secuenciación pasaron de secuenciar unas cuantas muestras o fragmentos, hasta análisis de miles o millones de fragmentos en un solo ensayo por medio de secuenciación de siguiente generación (NGS, por sus siglas en inglés: next generation sequencing), por ejemplo. Esta técnica revolucionó las ciencias biológicas debido a su rendimiento masivo, escalabilidad y velocidad, lo que permite investigaciones en una amplia variedad de aplicaciones y estudios en sistemas biológicos a un nivel nunca antes posible (van-Dijk y col., 2018).
Por un lado, se tienen las secuencias de las especies o especímenes que se quiere identificar o estudiar y, por el otro, se debe saber si fueron o no identificados antes y si dicha secuencia ya está reportada. Con la información anterior, ambas secuencias son comparadas o alineadas. En el caso de que ya estén publicadas las secuencias se puede recurrir a sitios en la web que cuentan con herramientas bioinformáticas, como el GenBank (Benson y col., 2013), o mediante el uso de programas bioinformáticos (ambas maneras se describen posteriormente).
Así, los análisis genómicos se basan en metodologías moleculares, las cuales muestran avances importantes en las técnicas de identificación de individuos o especímenes, lo que permiten detectarlos incluso sin tener evidencia física o visual del organismo (Andruszkiewicz y col., 2017b). Cabe señalar que estos estudios pueden dividirse en análisis con o sin amplificación de los ácidos nucleicos, llamados estudios de metabarcoding y metagenómicos, respectivamente.
En paralelo, las características de especies de vida silvestre u organismos que se cree extintos, escasos, escurridizos a simple vista o que no se pueden cultivar por métodos tradicionales, pueden inferirse mediante la detección de su DNA. A partir de este punto, el concepto de DNA ambiental (eDNA, por sus siglas en inglés: environmental DNA) emergió, describiéndose como el DNA que se puede obtener de muestras tomadas en el ambiente sin estar presente algún individuo físicamente (Taberlet y col., 2012; Ruppert y col., 2019), es decir, a partir de muestras de piel, pelo, fluidos u otros tejidos o materiales, como el suelo, aire o agua. Por lo que detectar el DNA y conocer a qué organismo o entidad biológica se parece es importante para obtener información sobre especies, poblaciones y comunidades (Thomsen y Willerslev, 2015) habitantes en un área específica o en determinado momento del tiempo.
Asegurar el éxito de la detección del eDNA dependerá de saber manejar la muestra recolectada y el posterior aislamiento e integridad del material genético obtenido de dicha muestra del medio ambiente (Andruszkiewicz y col., 2017a), hasta su identificación.
Este trabajo tuvo como objetivo dar a conocer el estado actual, usos y aplicaciones del estudio del eDNA, sus bondades, características, dificultades y aplicaciones en el estudio de la biodiversidad, salud, ambiente y de muestras antiguas.
I. eDNA
El término eDNA fue introducido en 1980, utilizado para detectar y describir comunidades de microorganismos en sedimentos marinos (Ivanova y col., 2019). En 1990 fue clasificado de acuerdo al tamaño de la partícula: DNA mayor a 0.2 μm fue descrito como DNA particulado, y menor a esto, DNA disuelto (Díaz-Ferguson y Moyer, 2014).
Para profundizar en el concepto: el DNA está contenido en materiales liberados por los organismos hacia el ambiente, los cuales pueden ser desde heces, mucosidades, células de la piel, organelos, gametos, inclusive, hasta DNA extracelular (Deiner y col., 2017). Como ya se demostró en diversos estudios, la técnica puede proporcionar información de macroorganismos extintos u organismos que hace tiempo transitaron por un punto dado pero que ya no están en el área. Esto se debe a que los materiales orgánicos conservan cadenas cortas de DNA que se mantienen en el ambiente por largos espacios de tiempo (Ficetola y col., 2008).
Sin embargo, la obtención de este tipo de DNA no es fácil, ya que hay factores ambientales que pudieran afectarlo (Hänfling y col., 2016), como la temperatura, el pH, la conductividad, o comunidades microbianas capaces de degradar el DNA (Andruszkiewicz y col., 2017a). Además, dependiendo del tipo de la muestra, como las heces, agua o sedimentos orgánicos, las concentraciones obtenidas en la extracción de DNA variarán, lo cual dependerá del espécimen, de su dieta, la estación del año, la densidad, que se encuentren en el agua o en el suelo, el tipo de sedimento (Valdez-Moreno y col., 2019), incluso de su pelaje. Los rangos de concentración de DNA que se puede obtener varían de 1 ng/μL a 10 ng/μL son suficientes para realizar un PCR de la muestra.
II. Bases técnicas del uso del eDNA
Para el estudio y análisis de eDNA se pueden seguir protocolos previamente publicados. En la metodología de Díaz-Ferguson y Moyer (2014) se argumenta que para la detección del eDNA se requiere el desarrollo de marcadores genéticos específicos para cada objetivo del trabajo. Los marcadores genéticos son secuencias conocidas dentro del ácido nucleico a estudiar y de las cuales se conoce normalmente su posición. Una vez detectados esos marcadores, se sintetizan sondas moleculares complementarias a ellos, para posteriormente ser empleados en diversos métodos, como el PCR, el cual amplifica millones de veces el fragmento de interés a estudiar, para enseguida ser visualizado a través de una electroforesis en geles de agarosa o poliacrilamida (Díaz-Ferguson y Moyer, 2014; Salipante y Jerome, 2020).
En algunas otras metodologías, como la que señalan Valdez-Moreno y col. (2019) y que se resume en la Figura 1, se utiliza el término metabarcoding, que implica la recolección de muestras ambientales (presencia física o no de los especímenes a estudiar), la extracción de DNA, amplificación por PCR para un gen de interés, por ejemplo, el gen mitocondrial de la citocromo oxidasa-I (COI por sus siglas en inglés: cytochrome c oxidase I) y secuenciación; también se puede emplear el procedimiento de “metagenómica”, para la cual no requiere de la visualización física del espécimen del medio ambiente y que, además, implica una colección de genes amplificados y secuenciados (pero sin pasar por un PCR, a diferencia del metabarcoding, que sí lo requiere). Es decir, mientras la metagenómica analiza secuencias nucleares, cloroplastídicas o mitocondriales diversas sin el uso previo de PCR para secuenciar, el metabarcoding se basa en el mismo principio pero se apoya del PCR y utiliza secuencias conservadas, universales y con regiones variables, que pueden servir como código de barras.
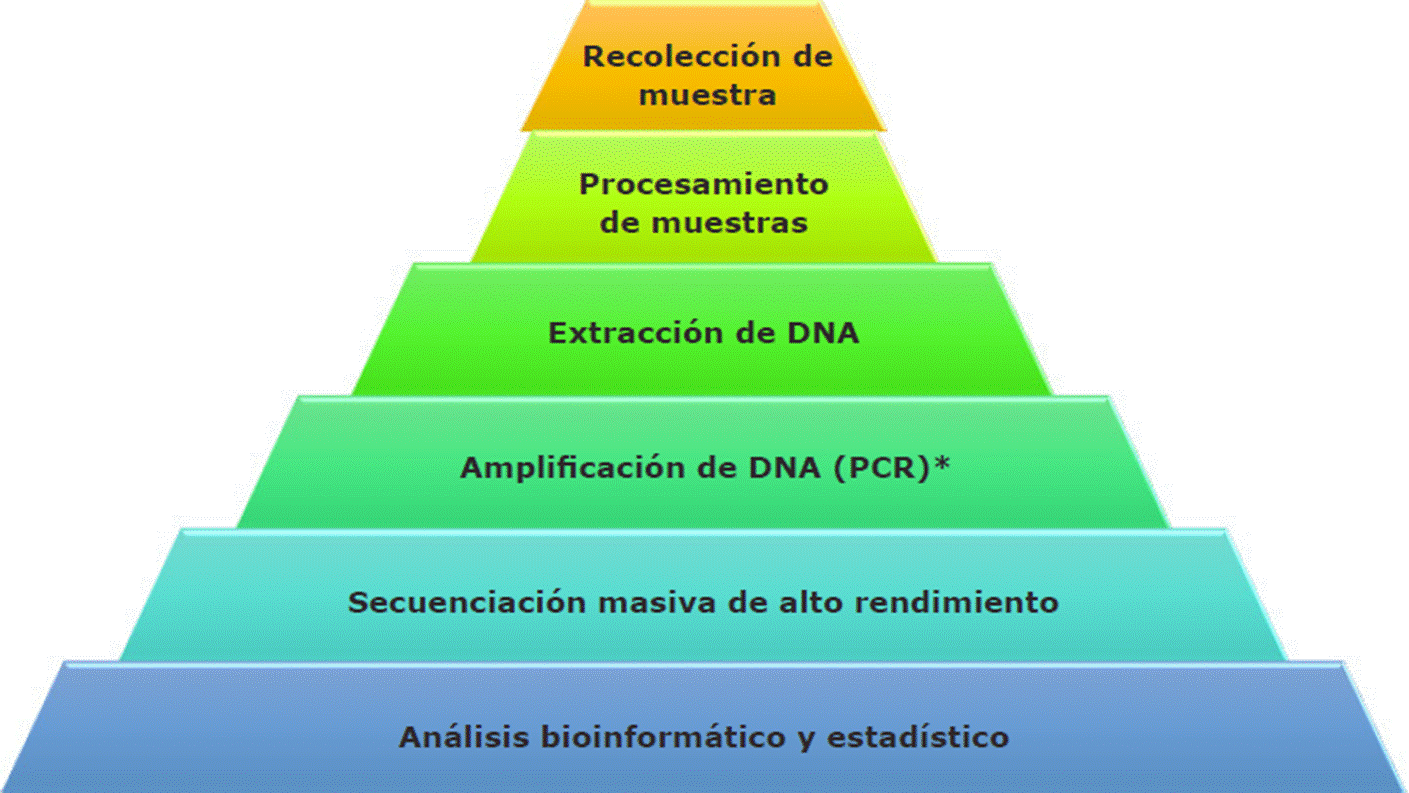
*Paso que puede o no ser omitido, dependiendo del tipo de análisis a realizar.
Figura 1 Diagrama de flujo del procedimiento general para la obtención y análisis del eDNA (Valdez-Moreno y col., 2019).
Figure 1 Flow chart for general procedure to extract and analyze eDNA (Valdez-Moreno y col., 2019).
Con lo anterior, se puede caracterizar la biodiversidad presente en una región dada, logrando la detección de especies o especímenes mediante la generación de sus secuencias específicas dentro del genoma o genoma mitocondrial del organismo, que servirán como una etiqueta molecular para la identificación de especies. Una serie de ejemplos de la versatilidad del estudio de eDNA se presenta como parte de la amplitud de este tipo de estudios. A modo de muestra, en el mar costero en Japón y en un río en Indiana, EE. UU., el DNA se aisló, se secuenció y se consiguió determinar, a partir de los restos de especímenes, el origen de una especie sin haber tenido conocimiento previo de su existencia en el lugar (Deiner y col., 2016; Lacoursière-Roussel y col., 2016; Valentini y col., 2016; Cowart y col., 2018; Valdez-Moreno y col., 2019).
Otros ejemplos de los usos de eDNA son en la eutrofización (el proceso de contaminación más importante de las aguas en lagos, balsas, ríos, embalses, entre otros) de las cuerpos de agua, tanto dulce como de mar, la cual es causada por los efectos de las actividades humanas y los aportes de nutrientes, lo que podría provocar la proliferación de algas nocivas (HAB, por sus siglas en inglés: harmful algal blooms), disminución de la calidad del agua, reducción de la biodiversidad, lo cual podría amenazar la salud humana (Liu y col., 2020); con la técnica podría identificarse los organismos presentes, benéficos o no y sus implicaciones en el ecosistema. Además, se podría estudiar el monitoreo de la biodiversidad, el eDNA podría usarse para estudios genéticos de poblaciones, lo cual es un campo emergente que ofrece perspectivas interesantes, sin embargo, habrá que seguir desarrollándolo (Adams y col., 2019).
Para obtener una muestra y su análisis como eDNA, a continuación se describen varios ejemplos donde se observa el uso de muestras de diferentes fuentes y orígenes, y su análisis.
II.1. Recolección de muestra de agua
Uno de los vehículos más comunes en el que se encuentra muestras de DNA o material con potencial DNA es el agua. De acuerdo a estudios realizados, donde se describe que la mayoría de los animales terrestres que se acercan a cuerpos de agua disponibles para beberla, tienen contacto de su saliva, tejido de su hocico o pelo y que éstos pueden caer y encontrarse en dicho fluido, es importante describir el procedimiento de cómo detectarlos. Por lo que se señala en general, el procedimiento marca que las muestras se tomen de la zona litoral a no más de 0.5 m, para posteriormente almacenarse en botellas estériles, aunque la toma de la muestra se puede realizar directamente de agua dulce, salobre, subterránea y de cuerpos tanto lénticos como lóticos. Equipo y muestras deben ser manipulados utilizando guantes, para evitar contaminar (material con DNA o RNA de las personas que manejan la muestra, de bacterias del ambiente o de otras muestras cercanas) y colocarlas en hielo, para su posterior procesamiento. Todo el equipo de muestreo y filtrado se debe lavar con una solución de hipoclorito de sodio (llamada también lejía o cloro) comercial al 10 % antes de su uso. Las muestras de agua tienen que ser recolectadas en envases de polietileno de aproximadamente 500 mL. En paralelo, dos muestras testigos negativos (agua destilada) deben ser colectadas en los recipientes, para hacer el seguimiento de una potencial contaminación durante el muestreo y el transporte del agua (Ushio y col., 2017; Valdez-Moreno y col., 2019). En México no se requieren permisos oficiales para la colecta de este tipo de muestras, ya que son del ambiente y no de vertebrados o de especies protegidas por las leyes mexicanas, pero en caso de que se produzca una perturbación en el sitio de muestreo o que se utilicen, aprovechen o descarguen aguas residuales sí se requeriría de un permiso oficial de acuerdo al Diario Oficial de la Federación (DOF, 2020) o lo procedente de acuerdo con las leyes y regulaciones del país o región de que se trate.
II.2. Procesamiento de muestra de agua
Las muestras de agua se deberán filtrar dentro de las 7 h de colectadas, para evitar que se degrade el material genético. Los artículos de filtrado más utilizados son los filtros de fibra de vidrio y los de nitrato de celulosa, con diferentes tamaños de poros. Los de poros más grandes permiten mayores volúmenes de agua y los de poros más pequeños capturan más partículas, pero limitan el volumen y la velocidad de filtración (Muha y col., 2019). Por ejemplo, existen filtros circulares de membrana de nitrato de celulosa Whatman, con tamaños de poros de 0.45 μm y diámetro 47 mm, y filtros de fibra de vidrio de borosilicato Advantec GA55 con tamaño de poros de 0.6 μm y diámetro de 47 mm. Ya en el laboratorio o lugar donde se procesará la muestra, se debe trabajar en un área estéril y tomar medidas para evitar la contaminación ambiental, por lo que habrá que cambiarse los guantes entre cada muestra o manejo de muestra, usar cubrebocas y, de ser posible, diferentes cuartos, tanto para la extracción del DNA como para la amplificación. Al terminar, el material filtrado se almacena a - 18 °C (Cowart y col., 2018; Valdez-Moreno y col., 2019).
II.3. Extracción del DNA
Para la extracción del DNA se pueden utilizar estuches comerciales (MoBio) normalmente vendidos como PowerSoil® DNA Isolation Kit - Qiagen, especiales para muestras de suelo, con indicaciones del fabricante que deberán seguirse. Este método usa filtros, a diferencia de otros que no los requieren. El procedimiento no dura más de 10 min. Posteriormente se debe incubar a 4 ºC el DNA obtenido. Otros estuches comerciales (Muha y col., 2019) son DNA Qiagen DNeasy Blood & Tissue (protocolo para manchas de sangre seca), usado para múltiples muestras, como tejido o sangre total. Uno más es Nexttec (Nexttec Biotechnologie GmbH, Alemania), con mayores ventajas debido a que evita una posible contaminación del DNA y que es de un solo paso entre la digestión (ruptura de paredes celulares y proteínas mediante enzimas o reactivos químicos) de la muestra y la elución final del DNA. También se puede usar el NucleoSpin® Soil (Soliman y col., 2017), el cual usa filtros y tiene una alta eficiencia en la obtención del DNA. Derivado de este último estudio, se recomienda para la estandarización el uso de diferentes individuos y por triplicado la extracción de DNA, lo que podría ser una solución para alcanzar un mejor protocolo para los estudios de metagenómica del suelo. Finalmente, se pueden usar procedimientos tradicionales para la extracción de DNA, como el de fenol: cloroformo (Green y Sambrook, 2017).
II.4. Amplificación del DNA
Para el estudio de las muestras de DNA existen diferentes zonas blanco que habitualmente se usan como regiones (segmentos de DNA) de amplificación. Varios son los ejemplos: COI, para metazoos, y la cadena grande de bisfosfato de ribulosa carboxilasa (rcbL), para las plantas, son los estándares establecidos por el código de barras de la vida. Otras regiones usadas son el RNA ribosómico 12S o el 16S, que pueden ser más apropiadas para diferentes taxones; para hongos se emplea ITS (por sus siglas en inglés: internal transcribed spacer) y en bacterias el 16S. Para ciertos eucariotas también se usa el 18S rRNA (Epp y col., 2012; Taberlet y col., 2012; Gibson y col., 2015; Creer y col., 2016; Deiner y col., 2017; Hering y col., 2018).
Los iniciadores para el metabarcoding de eDNA (Tabla 1) deben ser lo suficientemente cortos como para amplificar muestras degradadas, idénticas pero variables entre especies y flanqueadas por regiones altamente conservadas (segmentos de DNA que conservan su secuencia intacta a través del tiempo) para amplificar una variedad de especies sin sacrificar la especificidad del grupo objetivo. Se debe tomar en cuenta que se requieren procesos para asegurar que el producto amplificado sea real o verdadero y no falsos negativos, por lo que se realizan múltiples réplicas de PCR (realizar la reacción múltiples veces) y de réplicas biológicas (colectar más de una vez las mismas muestras biológicas).
Tabla 1 Ejemplos de los 8 iniciadores más utilizados en el estudio de eDNA (Evans y col., 2016; Thomsen y col., 2016; Andruszkiewicz y col., 2017b; Bylemans y col., 2018; Wang y col., 2021).
Table 1 Examples for the 8 commonly used primers in eDNA’s study (Evans y col., 2016; Thomsen y col., 2016; Andruszkiewicz y col., 2017b; Bylemans y col., 2018; Wang y col., 2021).
| Nombre iniciador | Sentido 5’ a 3’ | Secuencia | Amplicón (pb) |
|---|---|---|---|
| FishCBa | Directo | TCCTTTTGAGGCGCTACAGT | 130 |
| Reverso | GGAATGCGAAGAATCGTGTT | ||
| 16S1b | Directo | CGAGAAGACCCTWTGGAGCTTIAG | 107 |
| Reveso | GGTCGCCCCAACCRAAG | ||
| Ac16sc | Directo | CCTTTTGCATCATGATTTAGC | 375 |
| Reverso | CAGGTGGCTGCTTTTAGGC | ||
| 16S2d | Directo | GACCCTATGGAGCTTTAGAC | 233 |
| Reverso | CGCTGTTATCCCTADRGTAACT | ||
| 16S-Fishe | Directo | AGCGYAATCACTTGTCTYTTAA | 233 |
| Reverso | CRBGGTCGCCCCAACCRAA | ||
| Ac12sc | Directo | ACTGGGATTAGATACCCCACTATG | 429 |
| Reverso | GAGAGTGACGGGCGGTGT | ||
| MiFish-Uf | Directo | GTCGGTAAAACTCGTGCCAGC | 219 |
| Reverso | CATAGTGGGGTATCTAATCCCAGTTTG | ||
| Teleog | Directo | ACACCGCCCGTCACTCT | 100 |
| Reverso | CTTCCGGTACACTTACCATG |
*W = A o T; I = inosina, R = A o G, D = A, G o T; Y = C o T, B = C, T o G.
Una metodología de amplificación que se usa con buenos resultados es realizar un PCR de dos pasos. En el primero se utilizan iniciadores convencionales y en el segundo, iniciadores de fusión con identificadores moleculares únicos (UMI, por sus siglas en inglés: unique molecular identifiers) o los códigos de barras moleculares (MBC, por sus siglas en inglés: molecular barcodes), que son secuencias de ácido nucleico cortas o “etiquetas” moleculares, útiles en los protocolos de preparación de bibliotecas de secuenciación de siguiente generación. Después de la primera ronda, los productos de PCR se diluyen, para pasar a la segunda ronda de PCR. Finalmente, se purifica el DNA y se secuencia.
II.5. Análisis Bioinformático
Una vez obtenidas las secuencias, se comparan, resguardan y analizan. El primer paso pudiera ser subir las secuencias a una biblioteca mundial de secuencias. Existen diferentes bases de datos, una de las más conocidas es la del código de barras de la vida (BOLD, por sus siglas en inglés: barcode of life bata system). En ésta, cada secuencia cargada es clasificada y almacenada, para posteriormente ser comparada con secuencias ya existentes en el sistema. Una utilidad de lo anterior puede ser la clasificación de grupos de individuos relacionados, lo cual es conocido como unidad taxonómica operacional (OTU, por sus siglas en inglés: operational taxonomic unit), es decir, las secuencias son agrupadas por similitud de secuencias de DNA de un gen marcador taxonómico específico. Otra base de datos es la que se encuentra en el GenBank, que indica estar diseñada para proporcionar y fomentar el acceso de la comunidad científica a la información de las secuencias de DNA más actualizadas y completas, y forma parte de lo que es el sitio web de El Centro Nacional de Información Biotecnológica (NCBI, por sus siglas en inglés: National Center for Biotechnology Information), que promueve la ciencia y la salud al proporcionar acceso a información biomédica y genómica. Ello ayudará a determinar si la secuencia problema secuenciada existe o no en dicha base de datos, si es una secuencia conocida, o es la primera vez que se detecta.
Por otro lado, derivado de la generación de cientos, miles o millones de secuencias por análisis, fueron creados sitios web para reunir tal información, como es el caso de mBRAVE (multiplex barcode research and visualization environment), una plataforma multiusuario que admite el almacenamiento, la validación, el análisis y la publicación de numerosos proyectos basados en instrumentos de secuenciación de alto rendimiento (HTS, por sus siglas en inglés: high-throughput sequencing). Este sistema se basa en la plataforma BOLD para respaldar la identificación y el descubrimiento de especies para los datos de HTS (Ratnasingham, 2019).
El procesamiento y la subida de los datos a ese tipo de plataforma de almacenamiento se pueden llevar a cabo por medio de diferentes paquetes informáticos. Entre otros, QIIME2 (por sus siglas en inglés: quantitative insights into microbial ecology version 2), DADA2 (por sus siglas en inglés: the divisive amplicon denoising algorithm) u OBITools, que es un conjunto de programas de Python (lenguaje bioinformático) desarrollados para simplificar la manipulación de archivos de secuencias (Boyer y col., 2016; Callahan y col., 2016; Macher y col., 2021).
QIIME2 puede transformar secuencias sin procesar en diagramas de barras taxonómicas, árboles filogenéticos, análisis de coordenadas principales y otras visualizaciones de la diversidad microbiana. Para DADA2 se incluyen herramientas o procedimientos sobre cómo filtrar tablas y cómo calcular y visualizar los análisis de ordenamiento y medición de la diversidad, para lo cual se introdujo un enfoque basado en modelos para corregir errores del amplicón sin construir OTU. Así, DADA2 es precedido de otro paquete informático basado en el programa de R, el cual es de código abierto (https://github.com/benjjneb/dada2) que extiende y mejora el algoritmo de su predecesor. Mientras que OBITools hace la asignación taxonómica de secuencias automáticamente contra una base de datos en línea. Tiene capacidad para filtrar y editar secuencias teniendo en cuenta la anotación taxonómica y ayuda a establecer líneas de análisis a medida para una amplia gama de aplicaciones de metabarcoding de DNA, incluidos estudios de biodiversidad o análisis de dietas.
III. eDNA en agua
III.1. eDNA en acuíferos
El método para obtener eDNA de acuíferos se efectuó primeramente en ambientes controlados, para observar si era viable la detección de especies, para posteriormente ser aplicado a ambientes naturales (Ficetola y col., 2008). El eDNA es, relativamente, una nueva herramienta para el monitoreo que se utiliza en ambientes acuíferos, ya que las partículas de DNA se distribuyen homogéneamente en el agua, a comparación del DNA que pudiera encontrarse en el suelo (Hänfling y col., 2016). La mayoría de los estudios sobre este tema se enfocan en peces y anfibios, pero esta herramienta se diversificó tanto que también se usa para identificar mamíferos, reptiles, artrópodos, gasterópodos y bivalvos (Dougherty y col., 2016).
La técnica del eDNA mostró su utilidad al ser empleada por primera vez para el monitoreo de peces que habitan la región de Bacalar (México) y a la vez detectar especies invasoras que pudieran afectar las playas. Se aplicó en agua y sedimentos, descubriendo que era más viable en agua, ya que fue capaz de localizar un mayor número de organismos (Valdez-Moreno y col., 2019). En este estudio se incluyeron 8 sitios de muestreo (1 con 3 localidades), la mayoría fueron examinadas en más de una ocasión, produciendo un total de 14 eventos de muestreo. Las muestras fueron colocadas en botellas, y tanto éstas como el equipo de muestreo se manipularon con guantes, para minimizar la contaminación con DNA humano. En cada sitio se hicieron tres tomas repetidas de agua y sedimento. Cada muestra se dividió en dos submuestras de 0.5 L que se filtraron a través de filtros separados de 0.22 μm. Un filtro se almacenó con 1 mL de solución PW1 (solución encontrada en el estuche comercial DNeasy® PowerWater® Kit - QIAGEN) con medio de trituración (cuentas que ayudan al molido de la muestra), mientras que la otra submuestra se colocó con medio de trituración en un tubo seco cubierto con papel de aluminio.
III.2. eDNA en ríos
Los ríos son invaluables para los estudios ambientales, ya que transportan la información genética del paisaje a lo largo de los tramos que atraviesan, así como los sedimentos, material orgánico y los nutrientes a su alrededor (Deiner y col., 2016). El eDNA es empleado para el conteo e identificación de poblaciones de peces, principalmente, mediante métodos no invasivos, en contraste con los invasivos usados anteriormente para esto, donde eran capturados e identificados morfológicamente, lo que no ayuda mucho a la salud del animal, ya que le provoca estrés y aumenta su depredación (Shaw y col., 2016).
El eDNA resiste en el agua a temperatura ambiente y puede cambiar su integridad de unos días a algunas semanas, de acuerdo a un trabajo de Pont y col. (2018), que reveló patrones cuantitativos de la biodiversidad de peces. También señaló que en las corrientes de los ríos la concentración de eDNA y su detectabilidad no solo dependen de las tasas de producción y degradación, sino también de la dilución, el transporte a través de la red fluvial, la deposición y la resuspensión. Este estudio usó como referencia uno previo de Valentini y col. (2016), el cual diseñó iniciadores que se probaron exhaustivamente in silico utilizando el programa ecoPCR (Ficetola y col., 2010) en: (i) una colección de todas las secuencias de DNA mitocondrial y (ii) en todas las secuencias de DNA disponibles públicamente. El programa ecoPCR es un PCR in silico que consiste en seleccionar en una base de datos las regiones que coinciden (es decir, que presentan similitud) con dos iniciadores de PCR. Dichas regiones deben localizarse en la secuencia seleccionada, de manera que permitan la amplificación por PCR, lo que fuerza la orientación relativa de las coincidencias y la distancia entre ellas.
Deiner y col. (2016), mostraron que el método de eDNA ayudó a detectar más especies en comparación con un método convencional llamado “kicknet” (que utiliza una red de malla cuadrada con un mango de poste a cada lado que se emplea para recoger macroinvertebrados acuáticos en una corriente). Utilizando solo kicknet se consiguieron de 17 a 24 familias, y combinándolo con eDNA se obtuvieron de 23 a 40 familias (Deiner y col., 2016; 2017). Existen otros trabajos reportados en Indiana, EE. UU., donde el método combinado ayudó a identificar especies que otros métodos pasaron por alto, en comparación con el método convencional (kicknet). Deiner y col. (2016; 2017) identificaron un promedio de 12 especies, entre las que se encontraron Semotilus atromaculatus, Lepomis cyanellus, Etheostoma nigrum, Cottus bairdii, Rhinichthys obtusus, Catostomus commersonii, Oncorhynchus mykiss, Salmo trutta, Amblopites rupestris, Micropterus dolomieu, Lepomis macrochinus, y Etheostoma caeruleum; y con kicknet y eDNA se obtuvieron 4 especies más; Ameiurus natalis, Cyprinus carpio, Umbra pygmaea y Micropterus salmoides, dando un promedio de 16 especies en 2013 (Olds y col., 2016; Valdez-Moreno y col., 2019). Finalmente, otro estudio en Japón usando eDNA, descubrió especies acuáticas raras, en peligro de extinción, y especies invasoras (Yamamoto y col., 2017).
IV. eDNA en ambientes terrestres
Los sedimentos son una fuente muy valiosa para el estudio de eDNA, el DNA puede permanecer desde periodos cortos hasta miles de años, dependiendo de las condiciones del ambiente donde se encuentre la muestra (Buxton y col., 2018). Algunos estudios demuestran que el eDNA se mantiene más tiempo en el sedimento que en la superficie del agua. Turner y col. (2015) demostraron que el DNA de la carpa asiática de cabeza grande se conserva por 4 meses en el sedimentos y está más concentrado que en las columnas de agua (Turner y col., 2015).
Así como en el agua, los sedimentos terrestres son muy importantes, ya que ayudan a investigar y reconstruir evidencias de animales o comunidades de plantas que habitaron la tierra hace miles de años (Deiner y col., 2017). Es decir, la fauna y la flora ancestrales dejaron ahí rastros de DNA extracelular. En 2013, otro trabajo estudió sedimentos en Siberia y en Nueva Zelanda, descubriendo que contenían DNA de animales extintos, como el mamut lanudo, en el primer sitio, y las aves de Moa en el segundo (Pedersen y col., 2015). Para lograr el metabarcoding se utilizaron iniciadores genéricos (o universales), que están diseñados para apuntar a varios taxones simultáneamente, entre ellos, plantas, animales o muestras fecales, en contraste con los iniciadores específicos, diseñados para amplificar sólo unas pocas especies seleccionadas. Por esta razón y derivado, que con la tecnología molecular podía detectarse DNA, los investigadores le llamaron DNA ancestral o “antiguo” (aDNA, por sus siglas en inglés: ancient DNA).
Aunque parece “fácil” la detección del aDNA, se debe considerar que su campo está plagado de obstáculos metodológicos significativos que incluyen, entre otros, daño post mortem a biomoléculas conservadas, contaminación de muestras y reactivos por DNA moderno y la presencia de inhibidores de reacciones enzimáticas (McHugo y col., 2019). En general, diversos factores que pueden comprometer irrevocablemente la autenticidad y reproducibilidad de un DNA amplificado a partir de muestras arqueológicas o fortuitas encontradas en zonas donde posiblemente habitaron animales o plantas prehistóricas. Sin embargo, con los cuidados de muestreo, transportación, enriquecimiento del DNA en el momento de su extracción y, claro, la tecnología de la amplificación, se tiene el potencial para estudios de detección de flora y fauna ancestral.
V. eDNA en el aire
El agua y el aire son capaces de desplazar el DNA ambiental más rápido y a mayores distancias (Barnes y Turner, 2016). Específicamente, el aire es un medio importante de transporte de microorganismos ya que supera barreras geográficas (Yooseph y col., 2013) y traslada diferentes partículas de material diverso, polvo, microorganismos, esporas, polen, entre otros.
Por ello, el eDNA puede tener aplicaciones en la salud humana utilizando como muestra el aire ambiental de alguna zona o ecosistema dado. Determinar el microbioma en el aire puede influir en las decisiones de salud con respecto a los alérgenos (partícula o microorganismos que genera una reacción inmunológica de hipersensibilidad), los microorganismos patógenos y la contaminación del aire. Así se debe considerar, por ejemplo, la actual pandemia del síndrome respiratorio agudo grave (SARS, por sus siglas en inglés: severe acute respiratory syndrome), la cual es una enfermedad respiratoria viral causada por un coronavirus del tipo 2, llamado coronavirus asociado al SARS (SARS-CoV-2) que provoca la enfermedad conocida como COVID-19 (Wang y col., 2020). Para el caso de los microorganismos en aire, la identificación actualmente se realiza mediante la observación microscópica, la cual se torna laboriosa y en muchos casos no se logran reconocer, a nivel de género, todas las especies relevantes (Kraaijeveld y col., 2015) o cultivables, para su posterior identificación. Mediante la recolección de muestras del aire, aplicación del metabarcoding y la tecnología NGS es posible determinar la composición del microbioma en el aire y sus posibles implicaciones para la salud humana y el ecosistema.
Si bien, el uso más frecuente de metabarcoding ambiental es en entornos naturales, también se puede aplicar en interiores. En particular, los microorganismos transportados por el aire son una preocupación crítica en los espacios de atención a la salud, ya que algunos pueden causar infecciones asociadas a los hospitales y, en general, a lugares donde se tienen en uso equipos de aire acondicionado que pueden contener bacterias, virus u hongos (Cao y col., 2014; Banchi y col., 2018).
Un ejemplo de la aplicación de esta metodología fue en 2015, cuando se realizó un estudio en aguas marinas para la identificación de orcas (Orcinus orca); para ello se tomaron muestras de agua y aire/superficie de las Islas de San Juan. Del aire/superficie se obtuvo la muestra de DNA por medio de filtros. El análisis de las muestras de las superficie encontró excelente concentración de DNA y se logró secuenciar a los microorganismos (Baker y col., 2018). Para este estudio, además del eDNA, se utilizó una variante del PCR llamado PCR por gotitas digitales (ddPCR, por sus siglas en inglés: droplet-digital PCR), la cual es una nueva y poderosa tecnología para cuantificar niveles bajos de DNA mediante el fraccionamiento de una reacción de PCR en más de 20 000 gotas utilizando una emulsión de aceite (Doi y col., 2015).
En otro caso, se comprobó que se puede identificar el polen de las plantas que potencialmente es alérgeno. Lo que se hizo fue filtrar las muestras de aire (Kraaijeveld y col., 2015) para localizar el tipo de polen.
En realidad, la utilidad del metabarcoding de eDNA se vuelve infinita, en virtud de que es posible hacer un procedimiento de análisis relativamente sencillo, aislando un fluido (aire o agua) de uno o de diferentes entornos, y después determinando uno o varios organismos, con el examen del microbioma de un ambiente específico; en muchos casos, sin necesidad de crecimiento o siembra de tales microorganismos.
CONCLUSIONES
El avance de la ciencia conlleva a nuevos descubrimientos, técnicas y metodologías producto del trabajo de investigación, dedicación y tiempo. En biología molecular abarca nuevas maneras de analizar lo que antes era casi imposible, debido a que no se podían identificar simultáneamente a todos o una parte de los organismos de un ambiente dado. La reciente introducción de la técnica metabarcoding de ácido desoxirribonucleico (DNA) y en particular del estudio de DNA ambiental (eDNA) ha permitido alcanzar ese objetivo. Actualmente existe información disponible sobre el metabarcoding y eDNA, en particular, en agua, lo cual es muy relevante para la detección de especies nuevas, previamente descritas o especies invasoras para el ecosistema, los humanos, los animales o las plantas. Sin embargo, aún faltan más estudios en otros tipos de muestras, como suelos y aire. Se espera que en un futuro esta sea la manera para la caracterización y clasificación de especies y que su uso se generalice para el monitoreo de microorganismos, especies terrestres o acuáticas que se encuentran en números muy bajos, se cree que ya están extintas o son especies nuevas. Es importante destacar que se requiere de técnicas especiales, como la reacción en cadena de la polimerasa (PCR) y secuenciación de terminación de la cadena de Sanger o de siguiente generación. En esta primera etapa es necesario generar las bases de datos de todo tipo de especímenes, por ejemplo, de cuerpos de fluidos, para que existan referencias con qué compararse o en su defecto, si ya existen, comparar las secuencias obtenidas con esas bases de datos previamente creadas. Finalmente, con el metabarcoding se generará un código de barras para algún tipo de muestra que se desee estudiar y con esto lograr el inventario de la biodiversidad de un área en particular y por qué no, el inventario total de la Tierra, entre otras aplicaciones mencionadas en este trabajo.

