











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Chemical fixation of carbon dioxide (CO2) into useful chemicals has been drawing a huge of attention not only due to the fact that CO2 is the main greenhouse gas, but also CO2 is an abundant and nontoxic C1 building block for the synthesis of useful fine chemicals without any coproducts [1]. Currently, the direct fixation of CO2 by cycloaddition with epoxides is one of the most promising routes owing to important and versatile applications of the target cyclic carbonates in chemical industry [2]. In the past decades, numerous catalysts such as Lewis acids [3], transition metal complexes [4], organocatalysts [5], MOFs [6], mesoporous materials [7], CuCo2O4 [8], La2O3/TBAB [9], and others [10] have been studied for the synthesis of cyclic carbonates from CO2 and epoxides. However, most of these processes involve some draw back such as harsh reaction conditions, the use of cocatalysts (such as organic base or quaternary ammonium salt), limiting efficiency, low stability and applicability. Therefore, the development of effective and environmentally benign catalytic systems for the transformation of CO2 to cyclic carbonates under cocatalyst-free conditions is still an attractive topic.
Ionic liquids (ILs) have attracted much more attention owing to their unique properties, which have continued to grow diverse applications in fields of catalysis and environmentally benign chemistry [11]. Functionalized ILs have been demonstrated to be effective and environmentally benign catalysts for CO2 cycloaddition due to the synergistic effect of the functional cations and anions [12]. Despite it, these catalysts generally still suffer from drawbacks in costly ILs separation and recyclability. It is worth noting that the heterogeneous catalysts are more suitable for the operation, so the development of highly efficient and easily recyclable heterogeneous catalysts in combination with the characteristics of homogeneous ILs is highly desirable even in the absence of cocatalysts. The immobilization of functionalized ILs on porous materials supports have been made to develop new and efficient heterogeneous catalysts [13,14], which offer an alternative appealing strategy for the easily separated and reused of catalysts in chemical transformations.
Periodic mesoporous organosilicas (PMOs), as a class of mesoporous materials possess many properties in term of large specific surface areas, tunable pore sizes, heterogeneous distribution of functional groups, chemical and thermal stabilities, as well as highly ordered mesostructure, have been attracting great interests in catalysis and industrial chemistry [15]. Functionalized PMOs are new category of mesoporous organosilica materials with organic moieties in the pore wall and active functional groups in mesoporous channels, which are promising materials showing fast diffusion and inherent activity of functional groups. Moreover, functionalized PMOs offer a great alternative for high loading of the functional organic groups or the incorporation of metal species into silica wall framework and show potential and broad applications in heterogeneous separation and homogeneous catalytic reactions [16,17]. It is noteworthy that the introduction of functionalized ILs or metal complexes into the periodic mesoporous organosilicas forming ionic liquid- or metal-modified PMOs have been show promising catalytic performances [18]. Taking into account the excellent properties of periodic mesoporous organosilicas, should be of great interest to explore the use of these porous materials as supports to immobilize or incorporate active sites. Inspired by the outstanding catalytic properties of rare-earth metals in a variety of transformations [9,19], in this work we report the immobilization of functionalized benzotriazolium ionic liquid onto periodic mesoporous organosilica and further samarium acetate or lanthanum acetate incorporation for the design of novel and heterogeneous multifunctional La-/Sm-incorporating ionic liquid-based PMOs nanoparticles La-/Sm-PMO-ILCl, and then explored their use for catalytic transformation of CO2 to cyclic carbonates with epoxides under solvent- and cocatalyst-free conditions (Scheme 1). Furthermore, the heterogeneous supported catalysts exhibited an impressive catalytic activity in the cycloaddition as the intramolecular synergistic effect of the combination of rare earth sites of PMO and the functional groups of ILs accelerates the transformation.
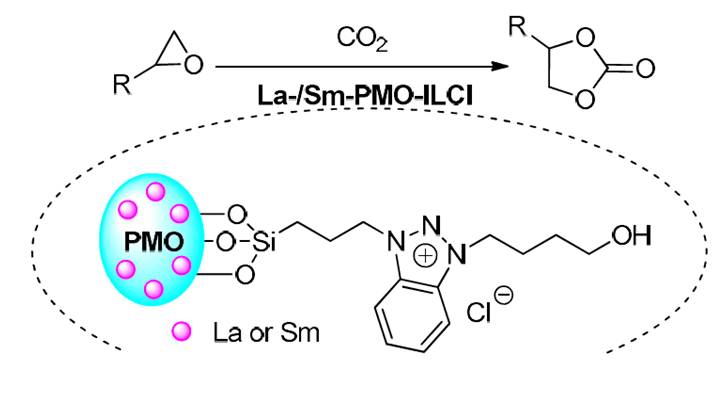
Experimental
Materials and apparatus
1,2-bis(triethoxysilyl)ethane and Pluronic 123 (EO20PO70EO20) were purchased from Sigma-Aldrich. The chemicals and reagents of analytical purity were obtained commercially without purification. FT-IR spectra were recorded on a PE Fourier Transform spectrometer recorded from 400 to 4000 cm-1 and potassium bromide as fixative. Scanning electron microscopy, energy dispersive X-ray spectroscopy (SEM, EDX) were performed on a JSM-7500F electron microscope,with the data recorded from 5 to 80° at a scan speed of 10°/min. Powder X-ray diffraction (XRD) analysis was recorded by Ultima IV diffractometer. UV-Vis spectra were recorded by Shimadzu UV-2450 spectrophotometer. Thermogravimetric analysis was measured by a NETZSCH STA 449 F5 apparatus with about 10 mg sample linearly heated from 30 to 600 °C (10°C/min). N2 adsorption-desorption isotherms were carried out using a BELSORP-max apparatus under liquid nitrogen atmosphere. NMR spectra were recorded on a Bruker 400 MHz spectrometer. Before being detected spectra, all the products were respectively diluted by CDCl3 reagent.
Catalyst preparation
Periodic mesoporous organosilica (PMO) was synthesized as described in literatures [15]. The multifunctional catalysts La-/Sm-PMO-ILCl were prepared using the similar synthesis routes as reported previously (Scheme 2) [13]. 1,2,3-benzotriazole (0.2 mol) and (3-chloropropyl) triethoxysilane (0.2 mol) was dissolved in 280 mL acetonitrile, and the reaction mixture was stirred at 80 °C under nitrogen for 24 h. After evaporating the solvent, the mixture was washed with ethanol (3 × 20 mL) and dried under vacuum to afford 1. Then, 4-chloro-1-butanol (0.1 mol), 1 (0.1 mol) and toluene (150 mL) were added to a round-bottomed flask, and the reaction mixture was heated to 100 oC under nitrogen for 24 h, the solvent was removed and the residue was washed with dichloromethane (3 × 15 mL) to afford 2. Subsequently, PMO-ILCl was synthesized in a one-pot reaction, PMO (1.0 g) and 2 (1.0 g) were added to a solution of dry toluene (80 mL), and the mixture was stirred and refluxed for 24 h under nitrogen. The resulting solid was collected by filtration and washed with acetone to remove excess IL. Then the above solid was dried under vacuum to afford the immobilized ionic liquid 3 PMO-ILCl. Finally, 3 (1.0 g), La(OAc)3 or Sm(OAc)3 (4 mmol), and acetonitrile (100 mL) were stirred at 80 °C for 24 h. After the reaction, the resulted material was collected by filtration, and then was washed with ethanol-water and dried under vacuum to afford the supported catalyst 4 La-PMO-ILCl or Sm-PMO-ILCl.
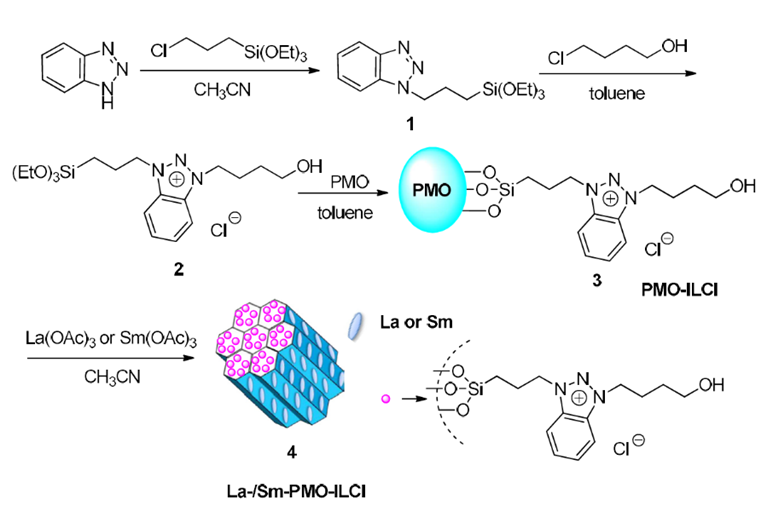
Catalytic cycloaddition of CO2 to epoxides
The cycloaddition was performed in a stainless-steel autoclave. In a typical reaction process, the reaction vessel was replaced with CO2, epoxide (10 mmol) and Sm-PMO-ILCl (0.4 g) without using any solvent and cocatalyst. At the desired reaction temperature, CO2 was introduced into the reactor to a fixed pressure and the reaction was stirred for a desired time. After the reaction was completed, the reactor was allowed to cool to ambient temperature and excess CO2 was released slowly. The resulting mixture was separated by centrifugation to remove the catalyst, and the product was analyzed by gas chromatography (Agilent 7890). Then the recovered catalyst was reused directly for the next run at the same cycloaddition procedures. The products were identified by comparing their physical and gas chromatography data with those of commercial compounds.
Spectra data of products
Propylene carbonate (Table 2, entry 1): 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.49 (dd, J=7.0 Hz, CH3, 3H), 3.96 (t, CH, 1H), 4.52 (t, CH, 1H), 4.81-4.86 (m, CH, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 19.4, 70.9, 73.6, 154.8.
Ethylene carbonate (Table 2, entry 2): 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.54 (s, CH2CH2, 4H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 64.8, 156.7.
1,2-Butylene glycol carbonate (Table 2, entry 3): 1H NMR (400 MHz, CDCl3): δ (ppm) = 0.93 (t, CH3, 3H), 1.56-1.62 (m, CH2, 2H), 4.41 (d, J=7.2 Hz, 2H); 4.59-4.63 (m, CH, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 12.9, 32.5, 69.2, 76.4, 154.8.
(Chloromethyl)ethylene carbonate (Table 2, entry 4): 1H NMR (400 MHz, CDCl3): δ (ppm) = 3.76 (dd, J=6.5 Hz, CH2, 2H), 4.35 (t, CH2, 1H), 4.57 (t, CH2, 1H), 4.93-4.98 (m, CH, 1H) ; 13C NMR (100 MHz, CDCl3): δ (ppm) = 43.7, 66.9, 74.4, 154.5.
Vinyl ethylene carbonate (Table 2, entry 5): 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.28 (dd, J=7.0 Hz, CH2, 2H), 4.93 (m, CH, 1H), 5.22 (t, CH2, 1H), 5.47 (t, CH2, 1H), 5.91-5.94 (m, CH, 1H) ; 13C NMR (100 MHz, CDCl3): δ (ppm) = 66.4, 76.7, 117.2, 135.7, 154.8.
Styrene carbonate (Table 2, entry 6): 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.35 (t, CH2, 1H), 4.76 (t, CH2, 1H), 5.68 (t, CH2, 1H), 7.31-7.42 (m, Ar-H, 5H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 70.7, 77.5, 125.9, 129.5, 129.7, 135.8, 154.9.
Results and discussion
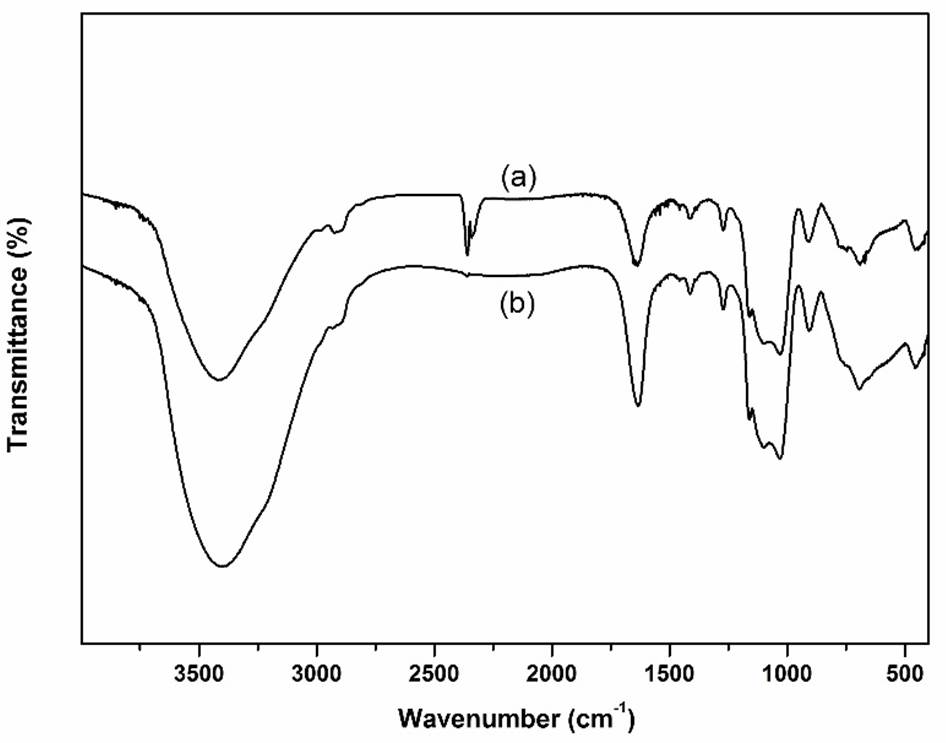
FT-IR spectra of the as-prepared supported catalysts are shown in Fig. 1. The typical adsorption peaks at around 1081 cm−1, 807 cm−1, and 489 cm−1, were attributed to the stretching vibration of Si-O-Si. The broad bands appeared about 3500-3350 cm-1 and 1642 cm-1 were assigned to the hydrogen-bonded stretching and bending vibration of supported catalysts [15-17]. The adsorption peaks appeared around 1172 cm-1, and 1032 cm-1 were ascribed to N=N, and C-N stretching vibrations of triazolium ring [20]. The absorption peaks appeared in the range 1459~1607 cm-1 and 3067 cm-1, were related to the stretching vibrations of aromatic benzene ring. The absorption peaks appeared around 2867 cm-1, 1296 cm-1, and 738 cm-1 were ascribed to the stretching vibrations of CH2 [14]. The characteristic absorption bands that appeared about 621 cm-1, 465 cm-1, and 473 cm-1 were ascribed to the C-Cl, La-O, and Sm-O vibrational modes, respectively [21]. These FT-IR results indicated the existence of typical functional species in the synthesized supported catalysts.
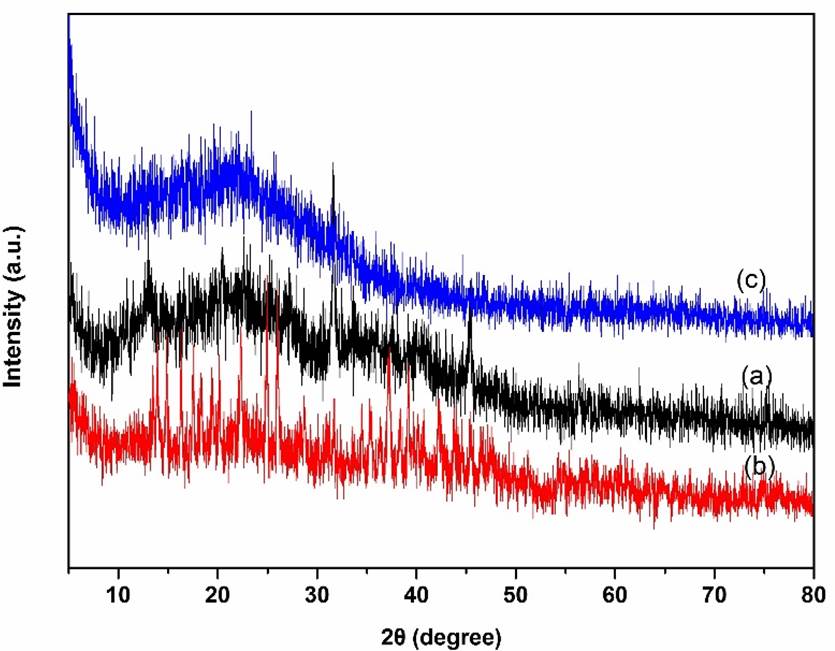
The XRD patterns for the as-prepared supported catalysts are presented in Fig. 2. The diffraction peak was observed at about 2θ = 23.2°, which is the characteristic amorphous phase of PMO [15,16]. The diffraction peaks at about 2θ = 25.6°, 27.9°, 30.4°, 39.5°, 46.1°, and 55.8° were attributed to the crystal planes of La nanoparticles. The peaks at about 2θ = 17.4°, 25.8°, 27.6°, 31.9°, 39.8°, 45.7°, and 60.5° compared to the parent PMO were attributed to the crystal planes of Sm nanoparticles [21]. No additional peaks corresponding to ionic liquids were observed, suggested that the ionic liquids species were well‐dispersed on the parent PMO framework. These results indicated that the basic amorphous phase of PMO framework maintained even after being immobilized of ionic liquids and then incorporated with rare earth complex.
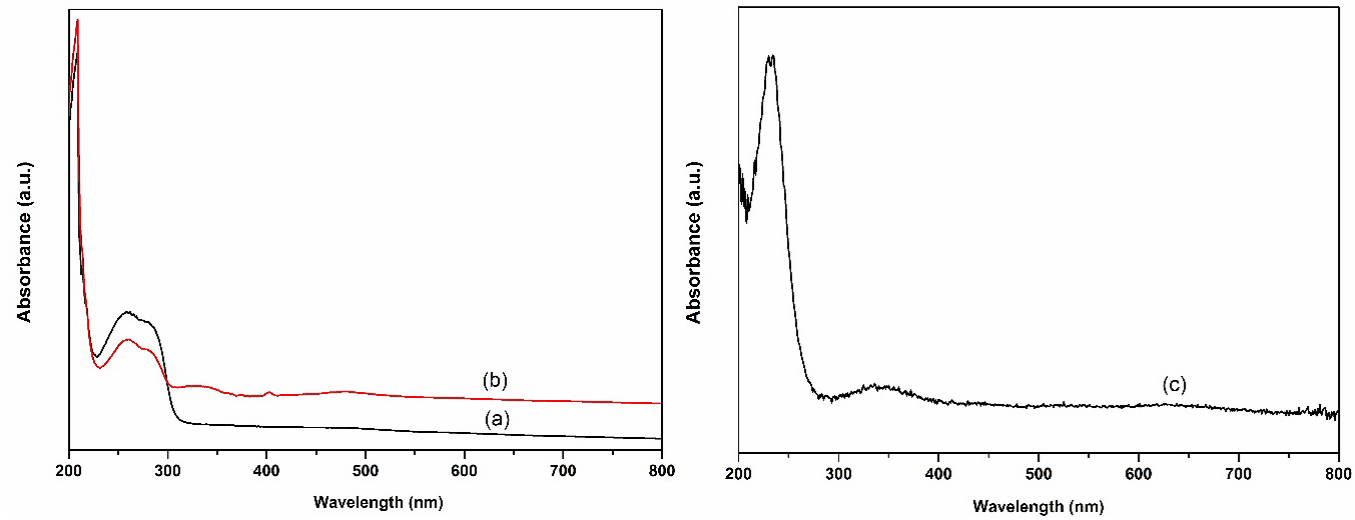
The supported catalysts were also characterized by UV-Vis (Fig. 3). The main absorption band at around 239 nm, corresponding to the characteristic Si-O bands absorption of PMOs [15]. It can be seen that, in the case of La-PMO-ILCl and Sm-PMO-ILCl, the absorption bands shifting to about 210 nm were ascribed to the electron transition of Si-O-La and Si-O-Sm. In addition, the peaks of immobilized ionic liquids in these supported catalysts were not observed.
The surface morphology of as-prepared supported catalysts was evaluated by SEM (Fig. 4). It can be seen from Fig. 4c the ordered rope-like structure, which was the characteristic morphological features of PMOs [15]. Figs. 4a,b demonstrated the SEM images of the La-PMO-ILCl and Sm-PMO-ILCl, which consisted of ordered rope-like structures, and their external diameter is about 7.45 nm. It can be clearly observed that PMO, La-PMO-ILCl and Sm-PMO-ILCl exhibited similar rope-like morphology, indicating that the immobilization of ionic liquids and subsequent incorporation of rare earth complexes do not nearly change the morphology of periodic mesoporous organosilicas. The elemental composition of as-prepared supported catalysts was investigated by EDX (Fig. 5 and Table 1). The EDX analysis suggested that there existed the expected elemental signals. The above analysis also revealed a pronounced decrease in the silicon and oxygen content in these nanomaterials. It was clearly observed that the carbon content of La-PMO-ILCl and Sm-PMO-ILCl could be increased significantly, which was due to the more carbon content of the grafted-IL precursor than PMO. In addition, after addition of rare earth complex, the peaks of samarium and lanthanum could be observed, and their content increased significantly. These results also successfully illustrated the well immobilization of ionic liquids species and incorporation of rare earth sites in the framework of PMO.

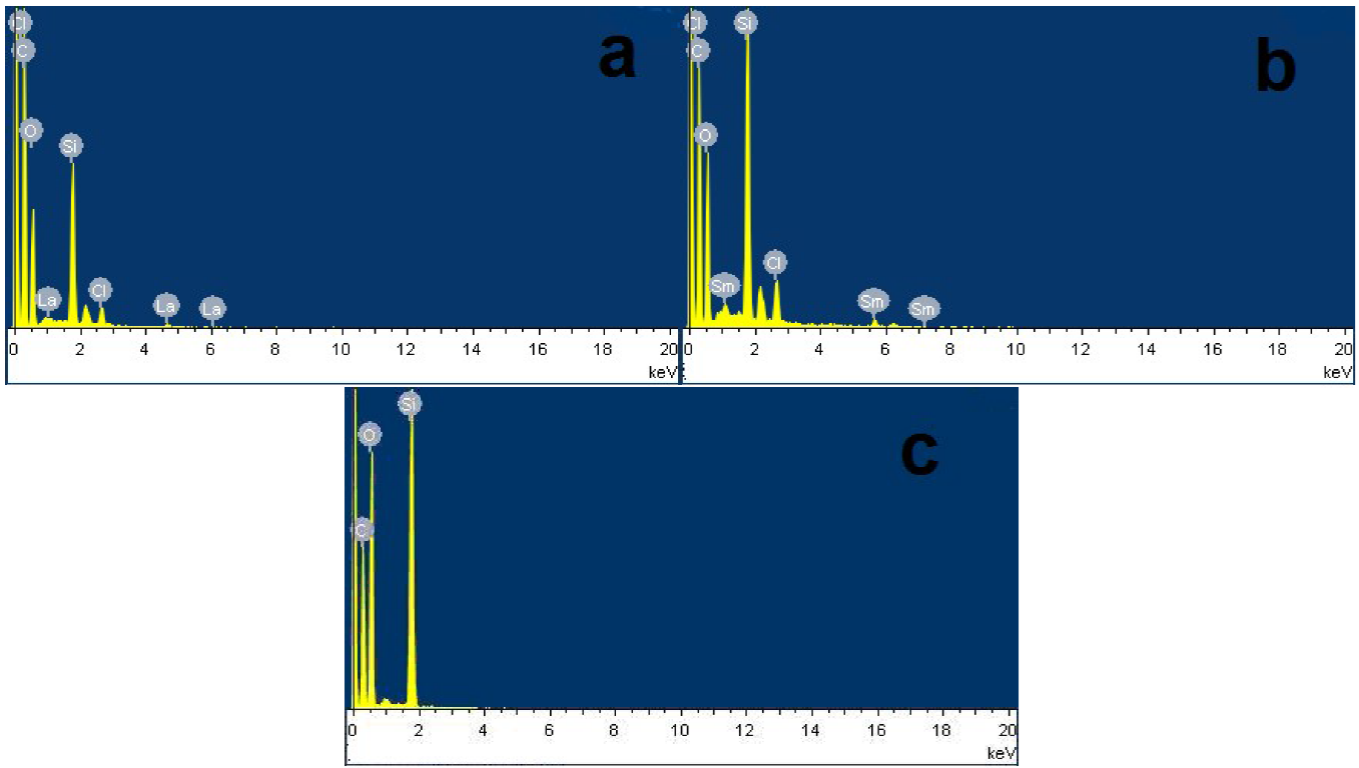
Table 1 EDX elemental composition of La-PMO-ILCl, Sm-PMO-ILCl and PMO.
| Sample | Si (wt.%) | C (wt.%) | O (wt.%) | La/Sm (wt.%) |
|---|---|---|---|---|
| PMO | 33.41 | 4.57 | 62.02 | - |
| La-PMO-ILCl | 28.79 | 10.89 | 46.93 | 7.96 |
| Sm-PMO-ILCl | 27.86 | 11.74 | 45.86 | 9.17 |
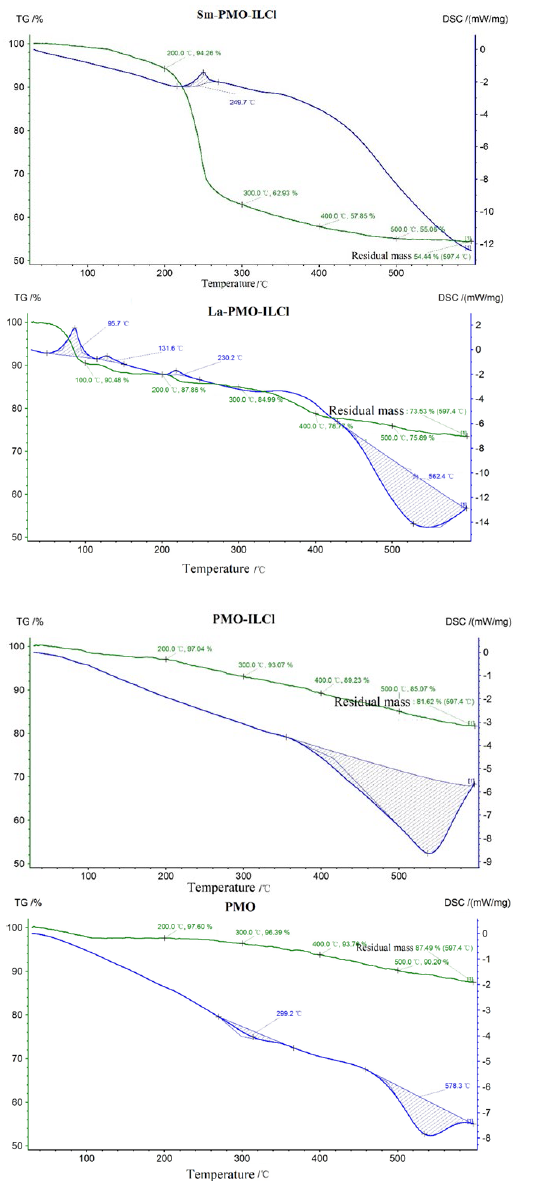
The N2 adsorption-desorption isotherms and pore size distributions of the as-prepared La-PMO-ILCl and Sm-PMO-ILCl are shown in Fig. 6. Similar to other mesoporous materials, the nanocatalyst exhibited the type IV isotherms with uniform mesoporous structure [15]. Following immobilization of ionic liquid and rare earth complex in PMO support displayed smaller surface area and pore volume, as well as slightly smaller pore size. It was clearly that the surface area, pore volume pore size and were reduced from 571.59 m2/g, 0.76 cm3/g and 7.19 nm for PMO to 353.49 m2/g, 0.53 cm3/g and 7.01 nm for La-PMO-ILCl, 286.38 m2/g, 0.40 cm3/g and 7.09 nm for Sm-PMO-ILCl, respectively, which was due to the immobilization of ionic liquid and rare earth active sites onto the mesoporous PMO. Furthermore, the pore size was still large enough for the freely diffusing of organic molecules. The thermal stability of Sm-PMO-ILCl was determined by thermal gravimetric analysis (TGA). Sm-PMO-ILCl depict a two main step thermal decomposition (Fig. 7). The first step of weight loss (5.74%) below 200 °C related to the removal of adsorbed water (≤100 °C) and other volatile solvent residues (100~200 °C), whereas the main weight loss (40.82%) from 200 to 600 °C in the second step was corresponded to degradation of the organic moieties of benzotriazolium ionic liquid. These observations demonstrated that the catalyst Sm-PMO-ILCl is thermally stable below 200 °C, which also indicated the successful immobilization of ionic liquid species and incorporation of samarium sites in the framework of PMO. Moreover, Sm-PMO-ILCl was observed to possess a large weight loss compared with La-PMO-ILCl, PMO-ILCl and PMO, suggesting that more ionic liquids and Sm complexes were grafted in this catalyst.
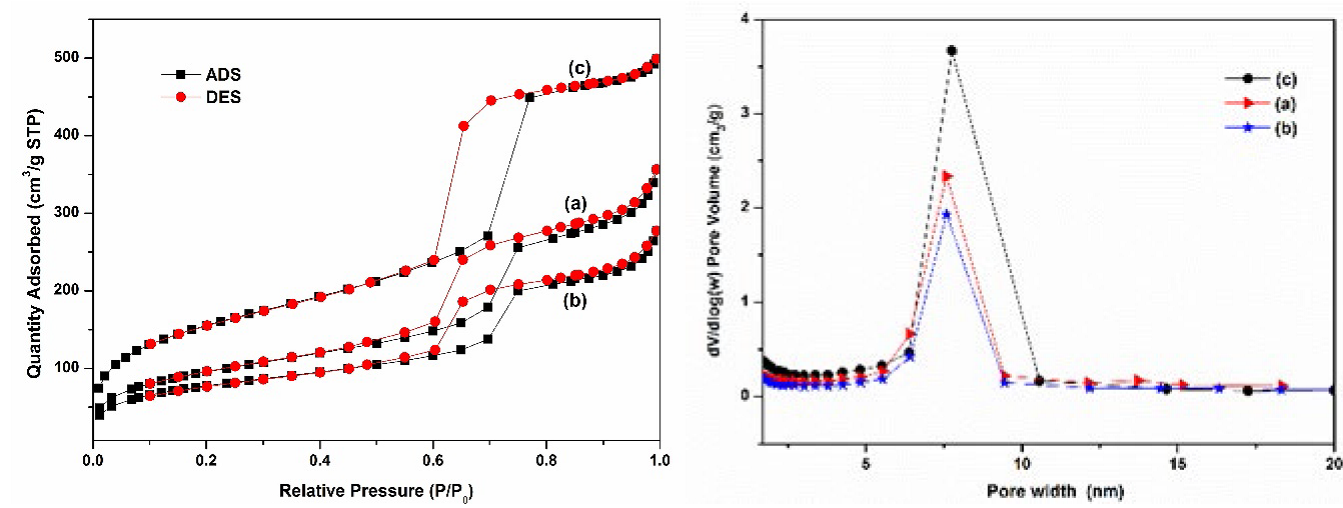
Fig. 6 N2 adsorption-desorption isotherms and pore size distributions of La-PMO-ILCl (a), Sm-PMO-ILCl (b), and PMO (c).
The well-characterized supported catalysts La-PMO-ILCl and Sm-PMO-ILCl were first screened for their catalytic performance in the cycloaddition of propylene oxide with CO2 to produce propylene carbonate as the model reaction (Table 2). Effects of the two supported catalysts were tested, it was found that the two supported catalysts La-PMO-ILCl and Sm-PMO-ILCl were effective catalysts, affording propylene carbonate with 89% and 97% yield, respectively, and the selectivity of propylene carbonate was ≥99% in all cases (Table 2, entries 1-2). The results showed that Sm-PMO-ILCl was the most effective catalyst with excellent selectivity of 99.6% for the cycloaddition in shorter time of 2 h (Table 2, entry 2). In contrast, no product was detected when the reaction was performed without a catalyst (Table 2, entry 6), the cycloaddition reactions were also carried out with the bulk IL, PMO support or PMO-ILCl as catalyst, resulting in low yields of 34%, 21%, and 75%, respectively (Table 2, entries 3-5). Thus, Sm-PMO-ILCl was identified as the proper catalyst in the next investigations considering all factors. The catalytic results revealed that the supported catalyst Sm-PMO-ILCl contributes to the enhancement of catalytic activity and selectivity in the cycloaddition, which may be attributed to the intramolecular synergistic effect from the samarium sites of PMO and the functional groups of ILs that accelerates the CO2 cycloaddition. Then, the effects of catalyst amount were also examined. The yield and selectivity were improved with increasing the catalyst amount (Table 2, entries 2, 7-9), while a further increase of the catalyst amount had little effect on reaction (Table 2, entry 10). Hence, the catalyst amount was found to be optimum at 0.4 g.
Table 2 Catalyst screening for CO2 cycloaddition to propylene oxide. a
| Entry | Catalyst | Catalyst (g) | Time (h) | Yield (%)b | Selectivity (%) |
|---|---|---|---|---|---|
| 1 | La-PMO-ILCl | 0.4 | 5 | 89 | 99.0 |
| 2 | Sm-PMO-ILCl | 0.4 | 2 | 97 | 99.6 |
| 3 | ILCl | 0.4c | 10 | 34 | 88.7 |
| 4 | PMO | 0.4c | 10 | 21 | 91.5 |
| 5 | PMO-ILCl | 0.4c | 7 | 75 | 97.3 |
| 6 | - | - | 24 | 0 | 0 |
| 7 | Sm-PMO-ILCl | 0.1 | 4 | 43 | 99.0 |
| 8 | Sm-PMO-ILCl | 0.2 | 3 | 74 | 99.1 |
| 9 | Sm-PMO-ILCl | 0.3 | 2 | 90 | 99.5 |
| 10 | Sm-PMO-ILCl | 0.5 | 2 | 97 | 99.5 |
a Reaction conditions: propylene oxide (10 mmol), CO2 (0.8 MPa), 100 °C. b Isolated yield, the selectivity of products based on GC analysis. c Reaction temperature at 120 °C.
The influence of CO2 pressure, and reaction temperature was then examined more closely. The influence of CO2 pressure was investigated as shown in Fig. 8, increased CO2 pressure from 0.2 MPa to 0.8 MPa resulted in an increase in the yield and selectivity of propylene carbonate. However, the yield and selectivity decreased as the CO2 pressure further increased from 0.8 MPa to 2.0 MPa. The initially increased the yield and selectivity may be due to the increased concentration of CO2 and oxide and propylene carbonate formed at high CO2 pressures. The subsequent decrease in the yield and selectivity beyond 0.8 MPa may be due to the reduction of propylene oxide concentration in the bottom catalyst phase, leading to the decrease in propylene carbonate product.4-6 Therefore, the appropriate CO2 pressure was 0.8 MPa. The influence of temperature was also studied and results are revealed in Fig. 9. It was observed that the yield and selectivity of propylene carbonate increased with an increase in reaction temperature up to 100 °C. However, further temperature increasing to 130 °C led to a decrease in the yield and selectivity, possibly due to the side reactions occurred at overly high temperatures, such as the isomerization and ring opening of propylene oxide (determined by GC analysis). Therefore, the optimum reaction temperature was 100 °C.
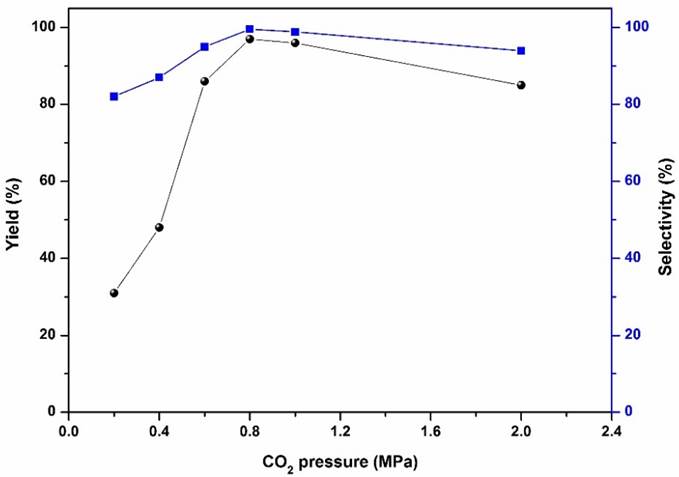
Fig. 8 Influence of CO2 pressure on the cycloaddition. Reaction conditions: propylene oxide (10 mmol), Sm-PMO-ILCl (0.4 g), 100 °C, 2 h.
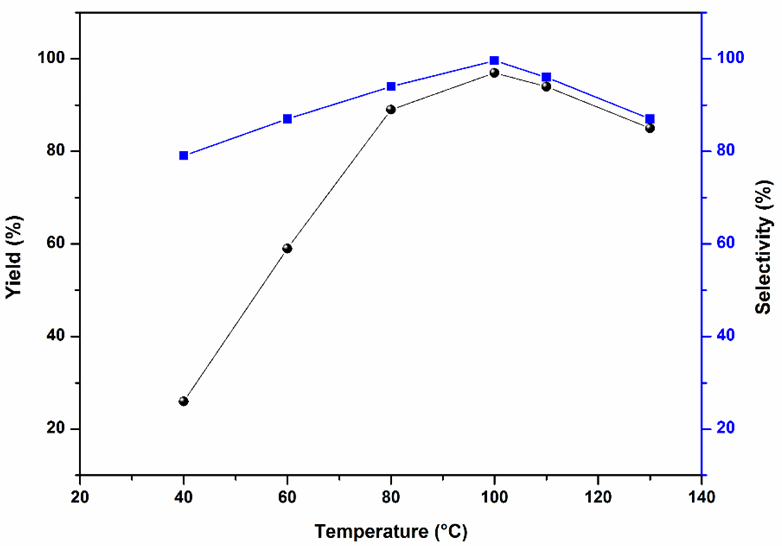
Fig. 9 Influence of reaction temperature on the cycloaddition. Reaction conditions: propylene oxide (10 mmol), Sm-PMO-ILCl (0.4 g), CO2 (0.8 MPa), 2 h.
In addition, recyclability of Sm-PMO-ILCl was studied in the cycloaddition to investigate the stability of the heterogeneous catalyst (Fig. 10). These results indicated that Sm-PMO-ILCl could be recycled and reused for at least five consecutive runs without significant losses in its catalytic activity. After each run, the catalyst was easily recovered from the reaction mixture by filtration and then reused directly for a subsequent similar reaction. The recovered catalyst after five runs was characterized by FT-IR, which showed similar spectra with fresh catalyst, indicating the stability of the catalyst (Fig. 11). Moreover, SEM image of the recovered catalyst after five runs showed similar morphology to that of the original one (Fig. 4d). Furthermore, a hot filtration test was performed (Fig. 12), the catalyst was filtered from the reaction mixture after 1.0 h. It shows that no reaction was proceeded in the filtrate after remove of catalyst, indicating that no obvious catalyst active sites leaching was observed. These results further confirmed that Sm-PMO-ILCl had excellent stability in the catalytic process.
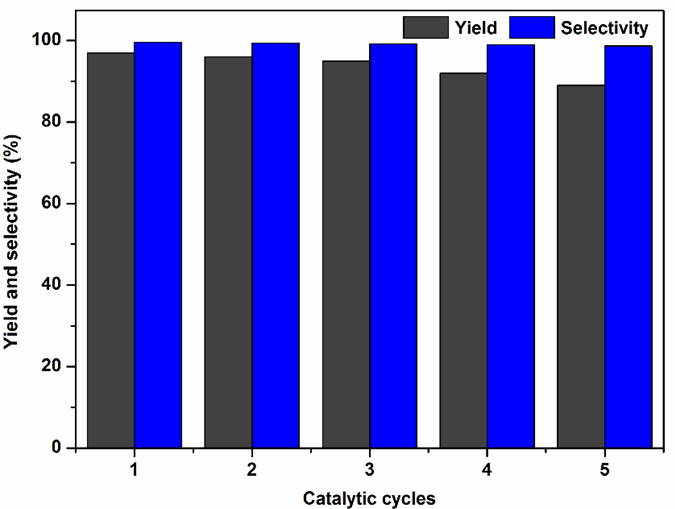
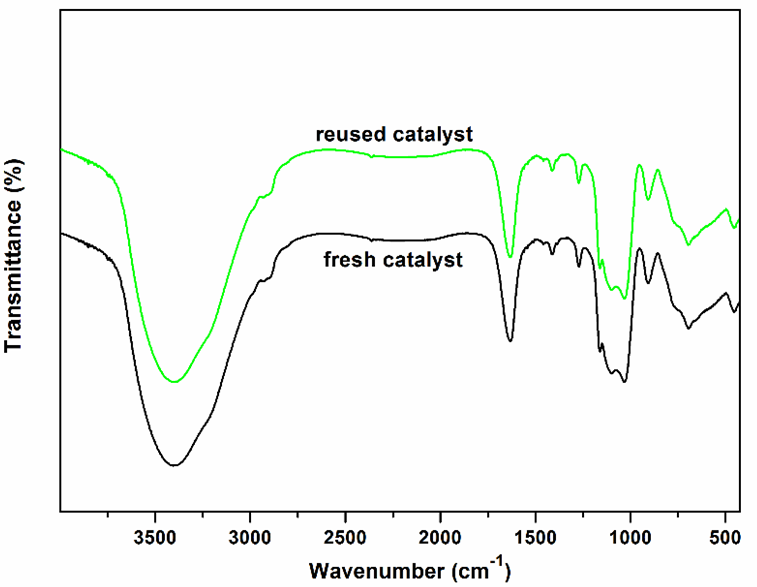
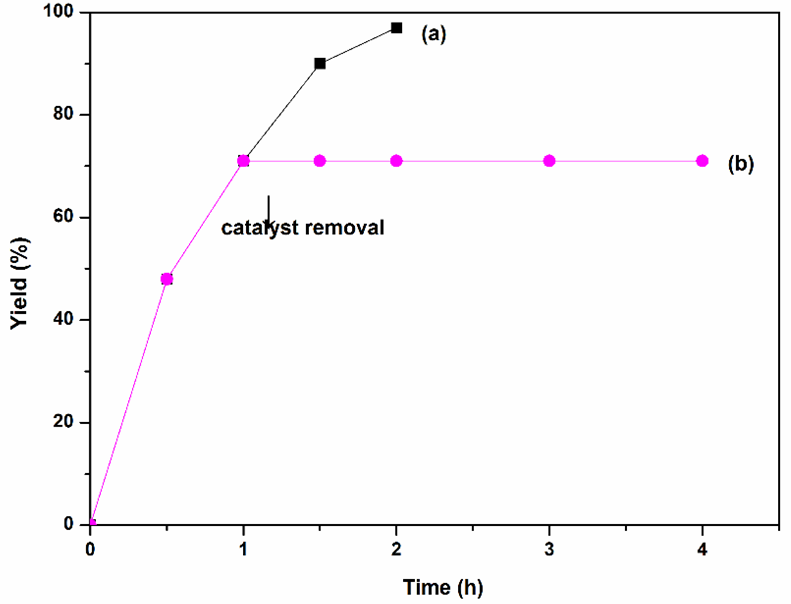
Fig. 12 Hot filtration test and leaching effect of Sm-PMO-ILCl in the cycloaddition. Reaction conditions: propylene oxide (10 mmol), CO2 (0.8 MPa) at 100 °C with catalyst (a) and catalyst removal (b) after 1.0 h.
Next, the scope in internal epoxide substrates was investigated to further evaluate the efficiency of the catalyst Sm-PMO-ILCl (Table 3). The cycloaddition reactions for different epoxides were examined, the protocol proceeded smoothly to give the corresponding cyclic carbonates in high yields and selectivities. Pleasingly, epoxides containing electron withdrawing and electron donating groups were converted to the desired products in 88-99% isolated yields. It was clear that oxirane was the most reactive epoxide (Table 3, entry 2), while the reactions of 2-vinyloxirane and 2-phenyloxirane requires longer times to obtain good yields (Table 3, entries 5 and 6). These results indicated the high efficiency and generality of Sm-PMO-ILCl.
Table 3 Catalytic cycloaddition of various epoxides with CO2 . a
| Entry | Epoxide | Product | Time (h) | Yield (%)b | Selectivity (%)b |
|---|---|---|---|---|---|
| 1 |
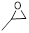
|

|
2 | 97 | 99.6 |
| 2 |
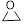
|
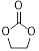
|
1 | 99 | 100 |
| 3 |

|
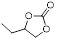
|
2 | 98 | 99.4 |
| 4 |
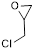
|

|
2 | 95 | 100 |
| 5 |

|
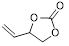
|
3 | 88 | 99.2 |
| 6 |
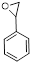
|
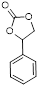
|
4 | 90 | 99.5 |
a Reaction conditions: epoxide (10 mmol), Sm-PMO-ILCl (0.4 g), CO2 (0.8 MPa), 100 °C.
b Isolated yield, the selectivity of products was > 99% in all cases based on GC analysis.
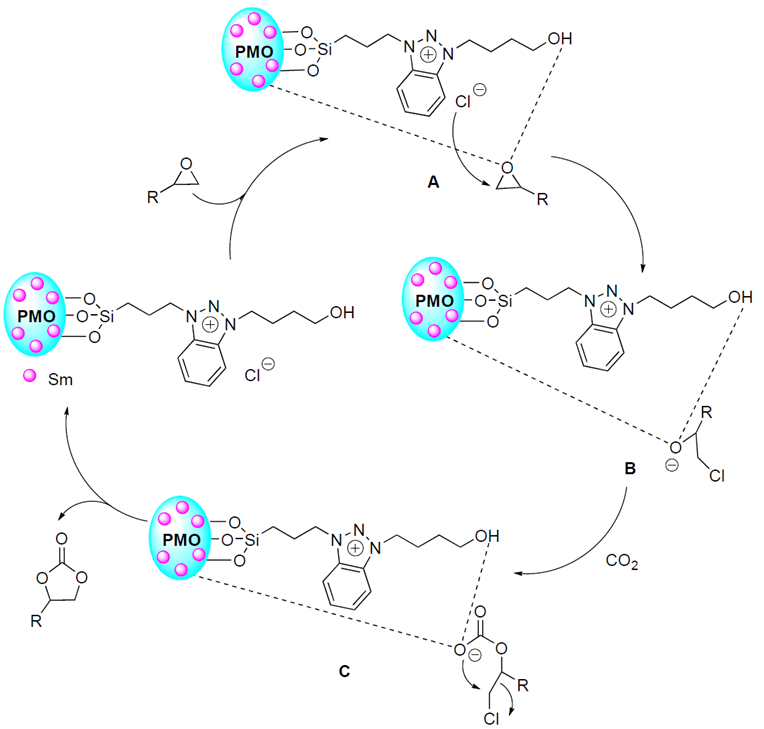
Based on the above results and previous reports [5,7-9], a possible mechanism was illustrated in Scheme 3. Initially, epoxide could be activated through the coordination interaction between samarium/hydroxyl active sites of Sm-PMO-ILCl and O atom of epoxide, resulting in the polarization of C-O bond, facilitating the formation intermediate A. Meanwhile, the nucleophilic attack of the Cl anion on the less sterically hindered carbon atom of epoxide generates the intermediate B. Then, there is nucleophilic interaction between the oxygen anion of intermediate B and CO2, forming the acyclic carbonate intermediate C. Finally, the desired cyclic carbonate can be obtained via intramolecular nucleophilic substitution of halide with simultaneous regeneration of the catalyst. The catalyst is recycled thereof and promotes the next catalytic cycle.
Conclusion
In conclusion, a novel type of well-ordered multifunctional periodic mesoporous organosilicas integrating immobilization of benzotriazolium ionic liquid and further incorporation of samarium acetate or lanthanum acetate were first afforded and characterized. The as-prepared La-/Sm-PMO-ILCl were presented as efficient catalysts for the cycloaddition of CO2 and epoxides to produce cyclic carbonates. Without any solvent and cocatalyst, the porous heterogeneous catalyst Sm-PMO-ILCl exhibited superior catalytic performance with ultra-high yields and selectivities in the cycloaddition reaction. Further, the catalyst displayed ease of recovery, excellent stability, recyclability and leach-resistance for at least five runs. The multifunctional Sm-PMO-ILCl intramolecular synergistic effect between samarium sites of periodic mesoporous organosilica and homogeneously dispersed basic sites of ionic liquid and thus provide sufficient catalytic sites during the catalytic cycloaddition. The proposed strategy in this work would offer interesting new potential for designing other highly efficient and environmentally benign catalytic processes.

