










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.80 no.2 Ciudad de México abr./jun. 2010
Artículo de opinión
Problemática de las cardiopatías congénitas en México. Propuesta de regionalización
Congenital heart disease in Mexico. Regionalization proposal
Juan Calderón–Colmenero, Jorge Luís Cervantes–Salazar, Pedro José Curi–Curi, Samuel Ramírez–Marroquín.
Departamento Cardiopatías Congénitas. Instituto Nacional de Cardiología Ignacio Chávez. México, D. F.
Correspondencia:
Juan Calderón Colmenero.
Instituto Nacional de Cardiología Ignacio Chávez,
Juan Badiano N°1. Col Sección XVI,
Tlalpan, 14080 México, D. F.
Correo electrónico: juanecalderon@yahoo.com.mx
Recibido el 29 de octubre de 2009.
Aceptado el 9 de febrero de 2010.
Resumen
Las malformaciones congénitas más frecuentes son las cardiopatías congénitas. La prevalencia reportada a nivel mundial va de 2.1 a 12.3 por 1000 recién nacidos. En nuestro país, se desconoce su prevalencia real; como causa de muerte infantil, se ubica en el sexto lugar en menores de un año y como la tercera causa en los niños entre uno y cuatro años; con base en la tasa de natalidad, se calcula que alrededor de 10 mil a 12 mil niños nacen con algún tipo de malformación cardiaca. Es de suma importancia conocer la magnitud del problema, identificar el número de niños que nacen cada año con una cardiopatía congénita y de manera desglosada por el tipo de la malformación; lo que permitiría determinar con mayor exactitud los recursos necesarios y planear su distribución. La regionalización tiene como objetivo la racionalización de los recursos con énfasis en servicios médicos muy especializados con la finalidad de lograr un mejor resultado para los pacientes. Por lo que, de manera simultánea a la creación de una base de datos fidedigna de las cardiopatías congénitas y, con base a la tasas de natalidad y mortalidad infantil secundaria a patología cardiovascular congénita y recursos en cada Estado, se debería proceder a regionalizar la atención. Lo anterior tendría diversos beneficios, ya que permitiría aumentar el número de casos atendidos, mejorar la calidad de la atención, aprovechar adecuadamente los recursos existentes y, seguramente, obtener una disminución de la mortalidad infantil.
Palabras clave: Cirugía cardiaca; Cardiopatías congénitas; Regionalización; México.
Abstract
Congenital cardiopathies are the most frequent congenital malformations. Reports of its prevalence around the world range from 2.1 to 12.3 for every 1000 newborns. Prevalence in our country remains unknown, but it probably occupies sixth place for mortality in infants less than a year old, and third place for mortality in those aged between 1 and 4 years. Based on birthrate, it is calculated that 10 to 12 000 infants in our country have some cardiac malformation.
To understand the magnitude of the problem, it is important to identify the global number of newborns with some congenital cardiopathy each year and the type of malformation that they have, in order to determine the necessary resources and to plan their distribution. The main objective of regionalization is the justification of the resources with an emphasis in the specialized medical services to provide the best results for the patients. Hence, reason, based on the resources of each state, as well as their natality and infant mortality rates related to congenital cardiovascular pathology, we should proceed to regionalize the attention, and to simultaneously create a trustworthy database of the congenital cardiopathies. This should have many benefits, such as increase the number of total attended cases, improve the quality of attention, use appropriately the existent resources, and –surely– decrease the infant mortality.
Key words: Cardiac surgery; Congenital heart disease; Regionalization; Mexico.
Introducción
En relación con las cardiopatías congénitas, consideramos necesario plantear varias preguntas: desde el punto de vista de Salud Pública, ¿qué importancia tienen en nuestro país?; a nivel nacional y en relación con otros sistemas de salud extranjeros, ¿con qué métodos de cuantificación de riesgo disponemos, que nos permita calificar nuestros resultados y compararnos?; ¿cuáles son las necesidades de recursos hospitalarios para una atención oportuna y de calidad? El análisis de los datos disponibles a nivel mundial, nos permitirá formular en este artículo, una propuesta de regionalización para el tratamiento de las cardiopatías congénitas en México.
Importancia
Está bien establecido que las cardiopatías congénitas, son las más frecuentes en el ámbito de las malformaciones al nacimiento.1,2 Para el propósito de este artículo, utilizamos como definición de cardiopatía congénita la de Mitchell y colaboradores,3 que habla de una anomalía estructural evidente del corazón o de los grandes vasos intratorácicos con una repercusión real o potencial. La prevalencia reportada de cardiopatías congénitas por 1000 recién nacidos vivos va de 2.1 en Nueva Inglaterra; de 2.17 en Estados Unidos y en Toronto, Canadá; de 8.6 en Navarra, España; de 10.6 en Japón y 12.3 en Florencia, Italia, por mencionar sólo algunos informes.2–5
Se desconoce la prevalencia real de las cardiopatías congénitas en nuestro país; la información de la que se dispone acerca de la importancia y repercusión de las malformaciones congénitas cardiacas se basa en las tasas de mortalidad que en 1990, las ubicaban en sexto lugar, como causa de muerte en los menores de un año, pasando a ocupar el cuarto en 2002; se constituye como la segunda causa de mortalidad a partir de 2005. En lo que corresponde a los niños entre uno y cuatro años, de ser la novena causa en 1990, escaló a la tercera en 2002 y se mantuvo en ese lugar desde 2005. La mortalidad total de la población pediátrica menor de 10 años fue de 15 548 pacientes desde 2004 hasta 2007, de los cuales, 83% corresponde a menores de un año (Tabla 1).

El diagnóstico de las cardiopatías congénitas ha sido posible por el advenimiento de métodos de diagnóstico que han facilitado su detección. Es el caso de la ecocardiografía, que ha permitido su detección en edades tan tempranas como la fetal y permitido identificar alteraciones anatómicas que antes requerían de la realización de un cateterismo cardiaco. También ha logrado caracterizar de manera más completa a las cardiopatías complejas así como las malformaciones cardiacas con mínima o nula sintomatología.6,7
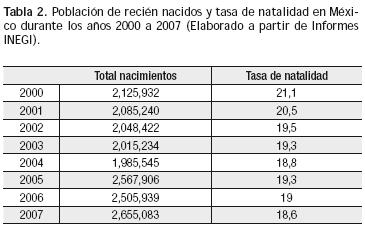
Al no disponer de la prevalencia real de las cardiopatías congénitas en nuestro país, puede considerarse un promedio teórico, derivado de la información mundial asequible: 8 por 1,000 nacidos vivos. Al relacionar esta cifra con la tasa de natalidad anual en nuestro país (2,500000); se puede inferir que cada año nacen alrededor de 18 mil a 21 mil niños con algún tipo de malformación cardiaca (Tabla 2).
Es importante conocer el número total de pacientes afectados con cardiopatías congénitas y saber la frecuencia de cada una de las variantes. Las cardiopatías congénitas tienen algunas diferencias regionales en su incidencia, pero en lo esencial, son muy similares en países Europeos, Estados Unidos y Canadá.
En un estudio realizado en la región de Bohemia, República Checa, que abarcó 10 años y 5,030 pacientes, las cardiopatías más frecuentes fueron: la comunicación interventricular (41%); comunicación interatrial (8.67%), seguido de estenosis aórtica (7.7%). Sólo otras cuatro cardiopatías más, tuvieron una frecuencia por arriba de 5%: estenosis pulmonar (5.8%); transposición de grandes arterias (5.3); coartación aórtica (5.2%) y persistencia del conducto arterioso (5.07%). Con menor frecuencia, se observaron los defectos de la tabicación atrioventricular (4%); síndrome de ventrículo izquierdo hipoplásico (3.42%) y tetralogía de Fallot (3.3%). La doble salida del ventrículo derecho, tronco común arterioso y atresia pulmonar con septo interventricular íntegro, representaron alrededor de 1%, cada uno. La conexión anómala total de venas pulmonares (0.8%); anomalía de Ebstein (0.4%); origen anómalo de coronarias (0.2%) e interrupción del arco aórtico (0.3%) tuvieron muy baja frecuencia.
Un análisis de 2257 pacientes con cardiopatía congénita realizado en el Hospital de Cardiología del Centro Médico Nacional Siglo XXI, mostró que la persistencia del conducto arterioso representó 20% de los casos, situación muy explicable por la altura a la que, con respecto al nivel del mar, está la Ciudad de México y zonas conurbadas; le siguió la comunicación interatrial (16.8%); comunicación interventricular (11%); tetralogía de Fallot y atresia pulmonar con comunicación interventricular (9.3%); coartación aórtica y estenosis pulmonar (3.6%) respectivamente y la conexión anómala total de venas pulmonares (3%). Es evidente que si se comparan los dos reportes sobre frecuencias de cardiopatías congénitas, aquellas que son secundarias a un defecto troncoconal, como la tetralogía de Fallot, son más frecuentes (9.3% vs 3.3%) y que la incidencia de conexión anómala total de venas pulmonares es también en forma significativa mayor (3% vs 0.8%). Ha sido mencionado que las cardiopatías por alteraciones troncoconales son más frecuentes en Japón con respecto a los Estados Unidos. Una situación similar es la encontrada en diversos estudios realizados en pacientes de países asiáticos, incluida India y países africanos, en los que la frecuencia de la tetralogía de Fallot, se da en el rango de 12% a 21%; asimismo, la frecuencia de la conexión anómala total de venas pulmonares es mayor con variaciones de 2.1% a 14.9%. Se ha establecido la posible similitud genética con los nativos de Asia lo que, en parte, podría explicar esta parecida frecuencia en tipos de cardiopatías congénitas.2, 8–14
Hoffman y colaboradores,15 reportaron que en Estados Unidos entre 1940 y 2002, nacieron 1.2 millones de niños con cardiopatía congénita catalogada como "sencilla", es decir pacientes que tenían una comunicación interventricular o un conducto arterioso de tamaño pequeño, estenosis pulmonar ligera, comunicación interatrial pequeña; 600 000 niños con una cardiopatía "moderada" donde se incluyó a pacientes con estenosis aórtica o pulmonar, coartación aórtica no crítica, comunicaciones interatriales amplias y finalmente, cerca de medio millón con una cardiopatía congénita catalogada como "compleja" y que incluyó defectos de la tabicación atrioventricular, formas complejas de comunicación interventricular y conductos arteriosos amplios, estenosis aórtica o pulmonar crítica, coartación aórtica severa. El porcentaje de supervivencia en niños atendidos con cardiopatía congénita simple o moderada fue de 75% a 80%; así como de 40% para los que tenían una cardiopatía compleja. En la actualidad, la supervivencia global de los recién nacidos con cardiopatía congénita, se sitúa en alrededor de 85%.
Boneva y colaboradores,16 describen una reducción de 39% en la mortalidad relacionada a cardiopatía congénita en el periodo de 1979 a 1997, lo que representó disminución de 2.5 a 1.5 por 100 000 habitantes. Ambos estudios, y otros más, confirman que con una atención oportuna y adecuada, se incide de manera evidente en la disminución de la mortalidad en los pacientes con cardiopatías congénitas.
Sistemas de evaluación del riesgo
En las últimas décadas se han desarrollado sistemas que tienen como fin estimar de una manera objetiva, la eficiencia y calidad de los servicios médicos. Se han agrupado pacientes con base al diagnóstico del tipo de cardiopatía para permitir la comparación en indicadores de calidad y costos, situación de suma importancia para las actividades hospitalarias.17
Para el tratamiento de las cardiopatías congénitas, existen alrededor de 140 procedimientos quirúrgicos, a los que hay que añadir los de cateterismo intervencionista, que las palian o corrigen. Por la elevada complejidad, producto del gran número de variantes de cardiopatías, el bajo volumen de muchas de ellas, así como la gran cantidad de procedimientos terapéuticos, ha sido difícil establecer tanto una nomenclatura como un sistema de estratificación de riesgos que pueda aceptarse con carácter de universal.18
En 2002 fue publicado un sistema de estratificación de riesgo, denominado Risk Adjustment in Congenital Heart Surgery (RACHS–1 por sus siglas en inglés), el cual plasma un consenso de once reconocidas autoridades médicas norteamericanas, clínicos y cirujanos, que se apoyaron en información de múltiples instituciones. Están incluidos 79 tipos de cirugía cardiaca y divididas en seis categorías de riesgo, la primera corresponde al menor riesgo quirúrgico (cierre de comunicación interatrial, ligadura de persistencia del conducto arterioso) y el sexto como el de máximo riesgo (cirugía de Norwood, como ejemplo).19
El RACHS–1 no fue pensado con el fin de predecir la mortalidad en un paciente determinado, sino como un sistema que permitiera comparar resultados quirúrgicos entre grupos de pacientes similares e Instituciones. El promedio de riesgo de mortalidad, referido para los diversos niveles corresponden a: riesgo 1: 0.4%; riesgo 2: 3.8%; riesgo 3: 8.5%; riesgo 4: 19.4% y riesgo 6: 47.7%. Por haber poca información, dado el escaso número de casos, no se ha podido estimar la mortalidad para el riesgo 5 (reparación de la válvula tricuspídea en pacientes neonatos con anomalía de Ebstein y corrección total de tronco arterioso común con interrupción del arco aórtico).20
Otro método de estratificación de riesgo fue publicado en 2004: sistema Aristóteles. En éste consenso intervinieron cirujanos cardiovasculares de 23 países y de alrededor de 50 Instituciones con objeto de evaluar la mortalidad hospitalaria. Además, también se enfatizó en definir más acuciosamente la complejidad de los diferentes procedimientos y de los pacientes.21 En este sistema existe el concepto de complejidad de un procedimiento quirúrgico, que se conforma por la suma de mortalidad operatoria: la ocurrida en los primeros 30 días; morbilidad: definida como el tiempo de estancia en cuidados intensivos postoperatorios y, finalmente; la dificultad técnica de la cirugía, dividida en cinco niveles que van del elemental hasta la muy difícil. El sistema cuenta con dos scores de puntaje: el básico y el completo. El básico se aplica a cada uno de los 145 procedimientos quirúrgicos, con una escala que va de 1.5 a 15 puntos. Estos procedimientos se agrupan en cuatro niveles de riesgo: 1: 1.5–5.9 puntos; 2: 6.0–7.9 puntos; 3: 8.0–9.9 puntos y 4: 10.0–15.0 puntos. El puntaje completo ajusta la complejidad con base en las características de los pacientes, y se dividen en dos grupos de factores: dependientes e independientes. En relación a los factores dependientes, el primer rubro considera variantes anatómicas, procedimientos asociados y la edad. Los factores independientes se dividen en generales: peso < 2.5 kg y prematurez; clínicos: que engloban variables presentes antes de la cirugía, (< 48 horas), e incluyen la presencia de acidosis metabólica (pH < 7.2); hiperlactatemia (lactato > 4 mmol/L); Disfunción miocárdica (FE < 25%); taquicardia ventricular; ventilación mecánica para manejo de la falla cardiaca; hipertensión pulmonar > 6 UW, por mencionar sólo algunos de ellos; extracardiacos: alteraciones cromosómicas o genéticas y quirúrgicos como la re–operación y la esternotomía de mínima invasión.
Tanto el sistema Aristóteles como el RACHS–1 son herramientas para permitir una autoevaluación y la comparación entre diversos centros especializados. Diversas investigaciones han tenido como objetivo validar ambos métodos. En el estudio realizado por Kang y colaboradores en Inglaterra para la validación del estudio de RACHS–1, abarcó 1085 cirugías consecutivas a corazón abierto, con una mortalidad global de 51 pacientes (4.7%). Evidenciaron que las variables independientes preoperatorias de mortalidad fueron edad (p < 0.002) y nivel de riesgo del RACHS–1 (p < 0.001) y transoperatoria el tiempo de circulación extracorpórea (p < 0.0001). Este grupo procedió a validar al mismo conjunto de pacientes de forma retrospectiva. El puntaje Aristóteles lo compararon con el RACHS–1, con lo que llegaron a la conclusión de que ambos sistemas son predictores de mortalidad (RA–CHS–1: p < 0.001; Aristóteles: p < 0.03).22, 23
En Nancy, Francia, se evaluaron ambos sistemas en una muestra de 201 pacientes que incluyeron tanto a pacientes pediátricos (164) como adultos (37), con una sobrevida de 97.56%. Llegaron a la conclusión de que el sistema Aristóteles permitía una mejor estratificación que el sistema RACHS–1.24
Holm Larsen y colaboradores,25 en Dinamarca, en un centro con un menor volumen quirúrgico que los anteriores, aplicaron la clasificación de RACHS–1 en pacientes atendidos de 1996 a diciembre 2002 con el fin de correlacionar la mortalidad y del periodo de estancia en sala de cuidados intensivos con los niveles de riesgo. La conclusión a la que llegaron es que la posibilidad de predecir la mortalidad hospitalaria fue similar a las referidas en Instituciones con un mayor volumen de cirugías y encontraron correlación directa entre nivel de RACHS–1 y la estancia en terapia intensiva.
Con la base de datos de 11 Instituciones donde laboran cirujanos miembros de la Sociedad de Cirujanos de Cardiopatías Congénitas (CHSS por sus siglas en inglés) se encontró que de los 16,800 procedimientos quirúrgicos realizados, 12,672 (76%) pudieron ser ubicados en los diversos niveles del sistema RACHS–1, la mortalidad general fue de 2.9%, con un descenso significativo con respecto al reporte de Jenkins y colaboradores, en los diferentes niveles de riesgo.7,26
Necesidad de recursos
Existen diversos estudios que fueron realizados con el fin de correlacionar el volumen de los casos manejados y los resultados clínicos. En 1979 Luft, Bunker y Enthoven,27 procedieron a analizar 12 procedimientos quirúrgicos en 1488 hospitales para determinar si existía relación entre el volumen quirúrgico y la mortalidad. Los autores encontraron que la mortalidad disminuía en hospitales que tenían un mayor volumen quirúrgico en cirugía a corazón abierto, cirugía vascular y revascularización coronaria.
En el campo de la cirugía cardiaca pediátrica, en 1995 Jenkins y colaboradores,28 realizaron el primer estudio que tenía como objetivo correlacionar el volumen quirúrgico y la mortalidad operatoria. En un grupo que abarcó 2833 niños operados de cardiopatía congénita en 37 centros hospitalarios de los estados de California y Massachussets, en los años 1988 y 1989. Dividieron los centros hospitalarios en varios grupos, de acuerdo al número de cirugías cardiacas pediátricas por año; aquellos que realizaban > 300 cirugías al año, los de un número entre 100 a 300 y, por último, aquellos centros en que se llevaban a cabo entre 10 y 100 cirugías. El resultado fue un descenso importante en la mortalidad operatoria en los centros hospitalarios con mayor volumen quirúrgico (> 300 cirugías por año).
Hannan y colaboradores,29 en 1998 publicaron una investigación que tuvo como fin el examinar la relación entre volumen de cirugía cardiaca pediátrica, tanto hospitalaria como por cada cirujano y relacionarlo con la mortalidad hospitalaria. El estudio incluyó 16 hospitales de Nueva York en el periodo entre 1992 y 1995. Se analizó un total de 7169 casos y se observó que en hospitales que llevaban a cabo un número < 100 cirugías cardiacas pediátricas por año, tenían tasas de mortalidad mayor (8.26%), en comparación con aquellos centros hospitalarios que tenían > 100 cirugías por año, cuya mortalidad fue de 5.95%. En cuanto a los cirujanos, se evidenció que los que realizaban al año < 75 cirugías resultaron con una mortalidad promedio de 8.77%, en comparación con los cirujanos que practicaban > 75 cirugías al año, cuya mortalidad fue de 5.9%.
Años después, Chang y Klitzner30 realizaron un estudio retrospectivo con información obtenida de 20 hospitales de California en los Estados Unidos, en la que se incluyó a 6592 niños que fueron sometidos a cirugía cardiaca entre 1995 y 1997, y llegaron a la conclusión de que la regionalización, es decir el reubicar a los pacientes de hospitales con bajo volumen a los de gran volumen quirúrgico, puede disminuir la mortalidad hospitalaria pero la reubicación incidió en los costos.
En un estudio realizado en 12 hospitales de Inglaterra en un periodo comprendido entre 1991 y 1995, se encontróque la mortalidad en niños mayores de un año fue 14.7% en hospitales de bajo volumen quirúrgico, con un promedio de 40 casos al año, en contraste con la mortalidad de 10% en centros con mayor volumen quirúrgico.31
Norwood y colaboradores, estudiaron los registros de 20 hospitales de Estados Unidos de Norteamérica para analizar los resultados quirúrgicos en relación a la corrección anatómica (procedimiento de Jatene) en la transposición de las grandes arterias encontrando que en los centros con menos de 10 operaciones al año la mortalidad fue de 55% y en los hospitales con más de 50 operaciones por año en promedio resultó de 9%.32
En 2005 fue reportadoun estudio por Checchia y colaboradores, 33que incluyó todos los pacientes menores de 30 días que ingresaron entre 1998 y 2001 con síndrome de ventrículo izquierdo hiipoplásico en 29 hospitales donde sellevaron a cabo 801 procedimientos de Norwood (estadio I) por 87 diferentes cirujanos. Se observó que en los hospitales donde se realizaba en promedio, por mes, una o más cirugías de Norwood tenían una sobrevida de 78% pero en aquellos hospitales que realizaban un procedimiento cada dos meses y medio o más la sobrevida disminuía a 59%.
En Suecia, hasta 1993, la cirugía de cardiopatías congénitas era realizada en cuatro ciudades, pero a partir de ese año se ubicó en los dos centros con menor mortalidad quirúrgica. Esta decisión obedeció a que la mortalidad quirúrgica, previo a este cambio, era de 9.5% (1988 a 1991) y fue reducida a 1.9% en 1995 a 1997, aun a pesar de que fue mayor la complejidad de las cardiopatías congénitas tratadas. Si bien la relación entre una menor mortalidad en los centros hospitalarios con un número elevado de cirugías cardiacas, ha sido establecida, no se cumple esta premisa en todos los casos, ya que existen hospitales y cirujanos con un número limitado de cirugías que reportan buenos resultados.34,35
La Academia Americana de Pediatría, en 1991, publicó las recomendaciones para centros que practican cirugía cardiaca y consideró que podrían ser rentables si: ofrecían atención a una población de 30,000 nacidos vivos cada año; si realizaran anualmente 100 cirugías cardiacas (de ellas 75% con circulación extracorpórea) y 150 cateterismos. En el 2002 la Academia Americana de Pediatría publicó nuevas guías, sin modificar esas cifras, pero resaltó la necesidad de participar en una red de salud regional y de mantener un adecuado número de casos que permita alcanzar resultados de alto nivel.36,37
El Comité de Cardiopatías Congénitas de la Asociación Europea de Cirujanos Cardiotorácicos (EACTS por sus siglas en inglés), dio la siguientes recomendaciones a las instituciones donde se lleva a cabo cirugía cardiaca: a) El número de pacientes intervenido por año debe ser de un mínimo de 250; b) En cuanto a recién nacidos y menores de un año el número de casos debe ser > 100 casos por año; c) Cada cirujano debe realizar un mínimo de tres intervenciones por semana y de 126 por año; d) en relación a unidades con un menor volumen de casos (< 250 pacientes intervenidos por año) se les puede considerar como Hospitales funcionales si los resultados son similares a los centros que manejan mayores volúmenes.38
Bases de la propuesta
De acuerdo con las recomendaciones establecidas por EACTS, debe haber un Centro Quirúrgico Cardiovascular por cada cuatro millones de habitantes; lo que implicaría que con una población en México calculada de 103 millones de habitantes, se requiere de alrededor de 25 centros de cirugía cardiaca para cardiopatías congénitas. Por otra parte, en México nacen al año cerca de dos millones de niños. Si nos apegáramos a los criterios de la Academia Americana de Pediatría, un centro médico–quirúrgico por cada 30 000 nacidos vivos, los requerimientos hospitalarios serían de 66 unidades de cirugía cardiaca.
El promedio europeo de intervenciones quirúrgicas por cada millón de habitantes es de 62, lo que extrapolado a nuestro país hablaría de la necesidad de 6386 intervenciones por año y, de acuerdo nuevamente a la recomendaciones de la EACTS, seria idóneo contar con 25 centros especializados.39 El promedio anual de cirugías de cardiopatías congénitas por año en España es de 51 por millón de habitantes, al aplicarlo a nuestro país, la necesidad de centros especializados sería de 21. Con base en estos criterios, para lograr resultados óptimos en la cirugía de cardiopatías congénitas en México, se requiere de 21 a 25 Centros Médico–Quirúrgicos Especializados.40
En nuestro medio existen 10 Centros Médico–Quirúrgicos Especializados en la atención para estos pacientes; ocho de ellos ubicados en la Ciudad de México, uno en la ciudad de Monterrey y otro en la ciudad de Guadalajara (Figura 1). Si bien es cierto que se realiza cirugía cardiaca de congénitos en otras ciudades del país y en hospitales privados, el número anual de cirugías no cubre los estándares mínimos recomendados en cuanto a volumen por hospital y cirujano cardiovascular, respectivamente.

La meta sería fortalecer en una primera fase 11 Centros Hospitalarios, en diferentes regiones del país para alcanzar un número mínimo de 21 sedes. Para considerar su ubicación habría que tomar en cuenta algunos aspectos como: a) la existencia de centros que cuenten con una infraestructura de tal nivel, que permita en un menor plazo fortalecerlos desde el punto de vista tecnológico y de recursos humanos; b) las regiones con una elevada tasa de natalidad o elevada tasa de mortalidad infantil secundaria a patología congénita cardiovascular; c) la gran dificultad en el traslado de los pacientes por diversos factores tanto orográficos como socioeconómicos y culturales.
La propuesta de distribución de los centros para la atención médico–quirúrgica de cardiopatías congénitas, se sustentó con base en la información de la base de datos de INEGI de la mortalidad por malformaciones del aparato circulatorio en menores de un año para el 2007 de cada uno de los estados del país, como se puede observar en las (Tabla 3). Con base en esa información y con las premisas arriba señaladas, la distribución propuesta para las once sedes podría ser la siguiente: Veracruz, Veracruz; Tuxtla Gutiérrez, Chiapas; Puebla, Puebla; Oaxaca, Oaxaca; León, Guanajuato; San Luís Potosí, San Luís Potosí; Morelia, Michoacán; Chihuahua, Chihuahua; Tijuana, Baja California Norte; Toluca, Estado de México; Mérida, Yucatán. Se pueden agregar cuatro más para alcanzar la meta de 25 Centros de Atención Especializada, considerando las ciudades de Culiacán, Sinaloa; Tampico, Tamaulipas; Torreón, Coahuila y por último Pachuca, Hidalgo (Figura 2).


Si tomamos en cuenta que en nuestro país prácticamente 50% de las cardiopatías congénitas están representadas por la persistencia del conducto arterioso, la comunicación interventricular e interatrial, éstas podrían ser resueltas, en una primera fase de la regionalización, en estos Centros de Alta Especialidad u Hospitales Estatales Pediátricos cercanos a la localidad donde vive el paciente, lo que podría reducir el costo familiar, social y disminuir de manera significativa la carga asistencial en los hospitales actuales de referencia, teniendo como objetivo alcanzar estándares de resultados aceptables de acuerdo a los métodos de evaluación RACHS–1 y Aristóteles. En su inicio y de manera ideal, estos Centros Regionales tendrían que ser apoyados con recursos humanos capacitados provenientes de los Centros de mayor experiencia hasta alcanzar resultados quirúrgicos adecuados de morbi–mortalidad.
Si además de las tres cardiopatías congénitas arriba señaladas, fueran manejadas la coartación aórtica no compleja y la fístula sistémico–pulmonar ya que, como se mencionó, la tetralogía de Fallot y la atresia pulmonar con comunicación interventricular con ramas pulmonares confluentes son muy frecuentes en nuestro país lo que permitiría alcanzar entre el 70 al 75% del total de las patologías congénitas cardiacas y, el porcentaje restante quedaría a cargo de los 10 Centros Especializados con los que se cuenta en la actualidad.
En una siguiente etapa, en los Centros Regionales de Especialidad para la atención de los niños con cardiopatía congénita, se procedería a realizar, de manera paulatina y previa evaluación de los estándares de calidad cirugías de mayor riesgo y complejidad incluidas la cirugía cardiovascular neonatal y derivando sólo pacientes con malformaciones cardiacas congénitas complejas como el síndrome de ventrículo izquierdo hipoplásico y cuyos resultados operatorios, en nuestro medio, son aun decepcionantes, entre otras razones, porque ningún centro dedicado a la atención de las cardiopatías congénitas en nuestro país, ha logrado reunir un volumen de casos suficiente para crear una sólida experiencia.
Conclusiones
La regionalización tiene como objetivo la racionalización de los recursos con énfasis en servicios médicos de alta especialidad con la finalidad de lograr un mejor resultado clínico para los pacientes. Con base en la población y los recursos existentes en cada Estado se debe proceder a regionalizar su atención y, mediante los métodos de estratificación de riesgo, evaluar de forma periódica para llevar a cabo adecuaciones al programa. Lo anterior tendría beneficios para la población ya que permitiría aumentar el número de casos atendidos, mejorar la calidad de la atención, obtener un óptimo aprovechamiento de los recursos existentes y, seguramente, lograr la disminución de la mortalidad.
Agradecimiento
Reconocemos la valiosa contribución de la doctora Ixchel Carranza Martínez en la elaboración de este artículo.
Referencias
1. Buendía A, Calderón CJ, Patiño BE, et al. Secuencia de estudio en el niño con cardiopatía congénita. PAC Pediatría I. México. Editorial Intersistemas. 2004:504–605. [ Links ]
2. Samanek M. Congenital heart malformations: prevalence, severity, survival and quality of life. Cardiol Young 2000;10:179–185. [ Links ]
3. Mitchell SC, Korones SB, Berrendees HW. Congenital heart disease in 56,109 births. Incident and natural history. Circulation 1971;43:323–332. [ Links ]
4. Fyler DC. Report of the New England regional infant cardiac program. Pediatrics 1980; 65:suppl:S376–S461. [ Links ]
5. Martínez OP, Romero C, Alzina AV. Incidencia de las cardiopatías congénitas en Navarra (1989–1998). Rev Esp Cardiol 2005;58(12):1428–1434. [ Links ]
6. Venegas C, Peña AY, Lozano R, et al. Mortalidad por defectos al nacimiento. Bol Med Hosp Infant Mex 2005 62:294–304. [ Links ]
7. Dirección General de Información en Salud, Secretaria de Salud. Estadísticas vitales en niños y adolescentes mexicanos. Mortalidad infantil. Bol Med Hosp Infant Mex 2004;61:515–527. [ Links ]
8. Alva EC. Lo esencial de la cardiología pediátrica. México DF. Ed. McGraw–Hill–Interamericana. 2006:73–81. [ Links ]
9. Bennerman CH, Mahalu W. Congenital heart disease in Zimbabwean children. Annals Tropical Paediatrics 1998;18:5–12. [ Links ]
10. Toefler OB. Congenital heart disease in aboriginals. Medical Journal of Australia 1979;1:620–630. [ Links ]
11. Van de Horst RL, Gorsman MS, Winship WS, et al. Congenital heart disease in South African Bantgu: a report of 117 cases. South African Medical Journal 1968; 42: 1271–1273. [ Links ]
12. Schrire V. Experience with congenital heart disease at Groote Schuur Hospital, Cape Town. South African Medical Journal 1963;37:1175–1180. [ Links ]
13. El Hag AI. Pattern of congenital heart disease in Sudanese children East African Medical Journal 1994;71:580–586. [ Links ]
14. Shann MKM. Congenital heart disease in Taiwan, Republic of China 1969. Circulation 1969;37:1175–1180. [ Links ]
15. Hoffman JI, Kaplan S. The incident of congenital heart disease. J Am Coll Cardiol 2002;39:1890–1900. [ Links ]
16. Boneva RS, Botto LD, Moore CA, Yang Q, Correa A, Erikson JD. Mortality associated with congenital heart defects in the United States. Trends and racial disparities. Circulation 2001;1003:2376–2381. [ Links ]
17. Jiménez J. Manual de gestión para jefes de servicios clínicos. Madrid. Editorial Díaz de Santos. 2000:430–435. [ Links ]
18. Mavroudis C, Jacobs JP. Congenital heart surgery nomenclature and database project: overview and minimum dataset. Ann Thorac Surg 2000;69:S1–S372. [ Links ]
19. Jenkins KJ, Gauvreau K, Newburger JW, et al. Consensus–based method for risk adjustment for surgery for congenital heart disease. J Thorac Cardiovasc Surg 2002;123:110–118. [ Links ]
20. Calderón–Colmenero J, Ramírez MS, Cervantes SJ. Métodos de estratificación de riesgo en la cirugía de cardiopatías congénitas. Arch Cardiol Méx 2008;78:60–67. [ Links ]
21. Lacour GF, Clarke D, Jacobs J, et al. The Aristotle score: a complexity–adjusted method to evaluate surgical result. Eur J Cardiothorac Surg 2004;25:911–924. [ Links ]
22. Kang N, Cole T, Tsang V, et al. Risk stratification in paediatric open–heart surgery. Eur J Cardiothorac Surg 2004;26:3–11. [ Links ]
23. Kang N, Cole T, Tsang V, et al. Does the Aristotle score predict outcome in congenital heart surgery?. Eur J Cardiothorac Surg 2006;29;986–988. [ Links ]
24. Macé L, Bertrand S, Lucron H, et al. Chirurgie cardiaque paedia–trique et autoavaluation: score de risqué; score de complexité et analyses graphiques. Arch Mal Coeur Vaiss 2005;98:477–484. [ Links ]
25. Holm LS, Pedersen J, Jacobsen J, et al. The RACHS–1 risk categories reflect mortality and length of stay in a Danish population of children operated for congenital heart disease. Eur J Cardiothorac Surg 2005;28:877–881. [ Links ]
26. Welke KF, Shen I, Ungerleider RM: Current assessment of mortality rates in congenital cardiac surgery. Ann Thorac Surg 2006;82:164–171. [ Links ]
27. Luft HS, Bunker JP, Enthoven AC: Should operations be regionalized? The empirical relation between surgical volume and mortality. N Engl J Med 1979; 301(25):1364–1369. [ Links ]
28. Jenkins KJ, Newburger JW, Lock JE, et al. In–Hospital Mortality for Surgical Repair of Congenital Heart Defects: Preliminary Observations of Variation by Hospital Case load. Pediatrics 1995;95:323–330. [ Links ]
29. Hannan EL, Raez M, Kavey RE, et al. Pediatric Cardiac Surgery: The effect of hospital and surgeon volume on In–hospital mortality. Pediatrics 1998;101:963–969. [ Links ]
30. Chang RKR, Klitzner TS. Can regionalization decrease the number of deaths for children who undergo cardiac surgery? A theoretical analysis. Pediatrics 2002;109(2):173–181. [ Links ]
31. Spiegelhalter DJ. Mortality and volume of cases in paediatric cardiac surgery: retrospective study based on routinely collected data. BMJ 2002;324:261–266. [ Links ]
32. Norwood WI, Dobell AR, Freed MD, et al. Intermediate results of the arterial switch repair: a 20–Institution study. J Thorac Cardiovasc Surg 1988;96:854–863. [ Links ]
33. Checchia PA, McCollegan J, Daher N, et al. The effect of surgical case volume on outcome after the Norwood procedure. J Thorac Cardiovasc Surg 2005;129:754–759. [ Links ]
34. Lundstrom NR, Berggren H, Bjorkhem G, et al. Centralization of Pediatric Heart Surgery in Sweden. Pediatric Cardiology 2004;21:353–357. [ Links ]
35. Mavroudis C, Jacobs JP. Congenital heart disease outcome analysis: methodology and rationale. J Thorac Cardiovasc Surg 2002;123:6–7. [ Links ]
36. American Academy of Pediatrics: Guidelines for Pediatric Cardiology Diagnostic and Treatment Centers. Pediatrics 1991;87:576–580. [ Links ]
37. American Academy of Pediatrics: Guidelines for Pediatric Cardiovascular Center. Pediatrics 2002:109:544–549. [ Links ]
38. Daenen W, Lacourt–Gayet F, Aberg T. Optimal structure of a congenital heart surgery department in Europe by EACTS congenital heart disease committee. Eur J Cardiothorac Surg 2003;24:334–351. [ Links ]
39. Monro JL. Surgery for congenital heart disease in Europe 1995. Eur J Cardiothorac Surg 1998;13:500–503. [ Links ]
40. Igual A, Saura E. Cirugía cardiovascular en España en el año 2001. Registro de intervenciones de cardiopatías congénitas. Cir Cardiov 2003;10:81–91. [ Links ]

