










Servicios Personalizados
Revista
Articulo
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de neurociencias (México, D.F.)
versión On-line ISSN 1028-5938versión impresa ISSN 0187-4705
Arch. Neurocien. (Mex., D.F.) vol.9 no.2 Ciudad de México jun. 2004
Arch Neurocien 2004; Vol. 9(2):85–93
ARTÍCULO DE REVISIÓN
Programmed cell death (apoptosis): the regulating mechanisms of cellular proliferation
JOSÉ ANTONIO MORALES GONZÁLEZ
Laboratorio de Bioquímica Médica, Facultad de Estudios Superiores Iztacala, UNAM, Los Reyes Iztacala, Estado de México.
Facultad de Odontología, UNAM, México.
AILEEN BUENO CARDOSO
Laboratorio de Bioquímica Médica, Facultad de Estudios Superiores Iztacala, UNAM, Los Reyes Iztacala, Estado de México.
FRANCISCO MARICHI RODRÍGUEZ
Facultad de Odontología, UNAM, México.
JOSÉ GUTIÉRREZ SALINAS
Laboratorio de Bioquímica y Medicina Experimental, División de Investigación Biomédica, Centro Médico Nacional 20 de Noviembre, ISSSTE.
Correspondence should be sent to:
José Antonio Morales González, MD, PhD,
Laboratorio de Bioquímica Médica, Facultad de Estudios Superiores Iztacala, Universidad Nacional Autónoma de México.
Av. de los Barrios No. 1. 54090 Los Reyes, Iztacala, Estado de México
E–mail: jmorales101@yahoo.com.mx
Recibido: 9 diciembre 2003
Aprobado: 19 diciembre 2003
RESUMEN
En la actualidad, la muerte celular que ocurre bajo condiciones fisiológicas es considerada como un fenómeno importante para lograr un desarrollo embrionario normal y mantener la homeostasis de tejidos y órganos del estado adulto. Este proceso de muerte está bajo control genético de manera semejante a los procesos de proliferación y diferenciación celular y, al igual que estos otros procesos, este tipo de muerte celular se presenta de manera predecible a lo largo del desarrollo. El hecho de que la muerte celular esté bajo el control de un programa genético específico, implica que la célula tiene una participación activa en su destrucción. Dadas las características morfológicas y bioquímicas de este tipo de muerte, se ha acuñado el término de apoptosis para distinguirlo de la muerte celular por necrosis. Los genes que controlan la muerte celular programada muestran un alto grado de conservación en diversos animales, comenzando con los nemátodos hasta llegar a los vertebrados superiores, lo que sugiere que este proceso apareció con el surgimiento de los animales multicelulares más primitivos. En este trabajo se revisan algunas de las características más sobresalientes de este proceso de muerte celular, a poco más de 25 años de su definición inicial.
PALABRA CLAVE: factor de necrosis tumoral, vale, caspasas, fragmentación internúcleo–somal del ADN.
ABSTRACT
The process of cell death under physiologic conditions has been recognized as an important phenomenon in normal embryonic development and in maintenance of tissue and organ homeostasis in the adult. This type of death is genetically controlled in a similar manner to the processes of cellular proliferation and differentiation and, similarly, the appearance of this type of cell death is predictable during development. The fact that a specific genetic program controls cell death implies that cells play an active role in their own destruction. Given the morphologic and biochemical characteristics of this type of death, the term apoptosis was coined to differentiate this process from necrosis. The genes that control programmed cell death show a high rate of conservation among various animals, beginning with nematodes all the way to higher vertebrates, suggesting that this process first appeared with the most primitive multicellular animals. Here we review some of the most relevant characteristics of this type of cell death nearly 25 years after it was first defined.
KEY WORDS: tumor necrosis factor, APO/FAS, caspases, internucleosomal DNA fragmentation
In 1972 Kerr, Wyllie, and Currie, while studying experimental systems of rodents, pointed out that the morphologic characteristics of natural cellular deaths during embryonic development, hepatic regeneration, and the balance between proliferation and death in in vivo tumor models in these experimental systems of rodents are different from those present in death as a result of pathologies or intoxication 1,2. On the suggestion of a colleague in the literature department, they began with the Greek word (that describes a leaf falling or the wilting of a flower) and coined the term apoptosis to describe this phenomenon, which has also been compared to cellular suicide. Although cellular death during embryonic development had been described previously, the contribution of Kerr, Wyllie and Currie proposed that in this process, cells destined to die participate actively in their our death and that this process is a determinant in maintenance of the cellular constitution of tissues 1,2.
Other studies on neuronal death during formation of functional synapses and on death of precursory cells of auto–reactive B and T cells during negative selection showed the importance of cellular death as a physiologic mechanism of selection 3,4. Later, genetic studies with the nematode Caenorhabditis elegans demonstrated the existence of genes whose expression controlled apoptotic death 5. Among genes identified were the gene ced–9 and by similarity in the sequence to its homolog in humans, the gene BCL–2, whose expression resulted in interference with death of neurotrophic factordepived neurons as well as with death of precursory cells of B and T 3,4–7.
To date, the accumulated evidence suggests that the great majority of animal cells are able to follow this path of death, even those in which the death program is not normally activated. This finding implies that this genetic program of self–destruction is part of the repertoire of cellular responses to external signals or changes in internal cellular conditions 8–10.
Throughout this revision, the terms programmed cellular death and death by apoptosis are used. Although they are used synonymously in the literature, they have different meanings. Programmed cellular death refers to the decision–making process; that leads to self–destruction of the cell. Death by apaptosis is a descriptive term that refers to the biochemical processes and morphologic changes that occur during cellular death and that are brought about by activation of the death program 2,4,6,11–13.
APOPTOSIS VERSUS NECROSIS
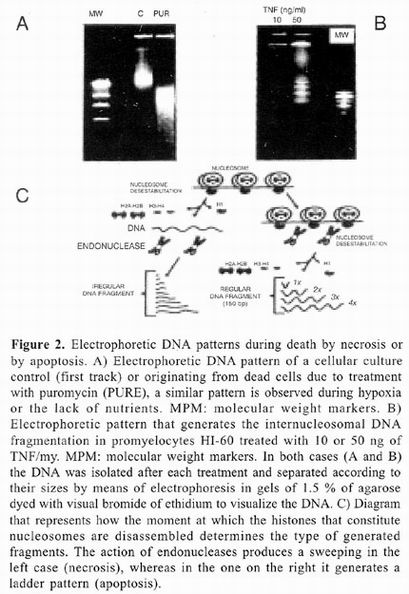
The biochemical and macromolecular processes that occur during death by apoptosis are clearly different from those that accompany death by necrosis. figure 1 outlines some characteristics of apoptotic death. Among the first morphologic changes displayed in cells that initiate the apoptotic process is condensation of cytoplasm and reduction of cellular volume, accompanied by changes in the structure of the nucleus (step 1 of figure 1). Chromatin is condensed and forms dense accumulations on the nuclear membrane. This is followed by invaginations of the nuclear membrane and ends with fragmentation of the nucleus into membranous structures with variable amounts of chromatin. In an analogous manner, the cellular membrane undergoes invaginations that end in fragmentation of the cell, forming clusters of variable sized vesicles containing intact organelles that do not merge with Iysosomes (step IV of figure 1). These vesicles are termed apoptotic bodies. In vitro, apoptotic bodies disintegrate, but in vivo they are quickly phagocyted by neighboring cells (step V of figure 1). The refore, one of the more relevant physiologic consequences of death by apoptosis is that intracellular material is not released into the interstitial space. It is important to note that this type of death is restricted to individual cells and never results in death of neighboring cells in which the death program was not activated 1–5.
On the other hand, cellular death by necrosis also affects not only the cell that dies, but also many adjacent cells. As during death by apoptosis, there are also nuclear chromatin aggregations responsible for darkening of the nucleus or pyknosis, wich nevertheless is less dense and less homogenous than that observed during death by apoptosis. At times, nuclear fragmentation can be also observed. Unlike during apoptosis, necrosis organelles are swollen and cellular volume increases considerably. These changes have been associated with alterations in osmotic control that results from depletion of ATP and consequent dysfunction of ionic membrane transports such the ATPase of Na+/K+. AII this leads to loss of membrane potential and increase in membrane permeability. Finally, membranes of cellular organelles merge and integrity of cellular membrane is lost, allowing cytoplasmic content to drain into the interstitial space 6–13.
Death by necrosis is generally accompanied by an inflammatory response followed by a healing process sometimes associated with development of fibrosis. A clear example of this type of death is hepatic tissue destruction as a result of cirrhosis or hepatitis. Unlike apoptosis, none of these changes is determined genetically and necrosis is rather the result of loss of functional synchronization between biochemical processes and macromolecular structures that constitute the cell 11–13.

APOPTOTIC DEATH DOES NOT EVOKE AN INFLAMMATORY RESPONSE
One of the characteristics of greater physiologic relevance of death by apoptosis resides in the fact that it does not induce an inflammatory response 1–5, 11–13. When a cell dies due to necrosis, its cytoplasmic content is spilled into the interstitial space exposing a great number of antigens that the immune system recognizes as foreign and to which it reacts attempting to eliminate them. During this process several cellular components of the immune system are activated, such as macrophages and neutrophils, wich release H2 02 damaging any cell with wich they have contact. The result is destruction of many cells in the surrounding tissue. It is important to note that formation of apoptotic bodies prevents release of cytoplasmic content into the interstitial space. Before apoptotic bodies are disintegrated, they are phagocyted by neighboring cells in a process in which surface antigens and phosphatidylserine participate, and that only under these circumstances appear in the external lamina of cellular membrane of apoptotic bodies 10–12. These molecules are recognized by specific receptors in collaboration with the major histocompatibility system. Compartmentalization of apoptotic cell content prevents intracellular antigens from evoking autoimmune reaction. This characteristic of cellular death by apoptosis is responsible for inflammatory response not taking place during tissue remodeling and reabsorption. Nonethless, these imply disappearance of complete organs and tissues, as in the metamorphosis of insects and amphibians 13 or reabsorption of embryonic Mullerian conduits in male vertebrates or mesonephros in females 10–14.
ACTIVATION AND INACTIVATION OF THE DEATH PROGRAM
Programmed cellular death is a selection mechanism in which microenvironment conditions surrounding a cell determine which cells must die and the moment at which this must occur. The study of different cellular systems in which apoptotic death takes place suggests the existence of two forms of activation of the death 1–3,16,17. In some types of cells, the death program is off, requiring an external stimulus to activate it (figure 1, diagram 1 a). Paracrine signals pertaining to the cytokine group, such as TNF or APO/FAS ligand, are prototype examples of signals that when united to their membrane receptors in target cells, activate the death 11,18,19. There are an increasing number of ligands and receptors recently identifted with a similar function. These systems allow for selective elimination of cells within a tissue. Whereas the APO/F AS system appears to be associated with elimination of T and B Iymphocyte clones that respond to their own antigens, the death system dependent on TNF participates in the death of tumor–like cells or cells infected with different types of virus.
Nevertheless, activation of the program does not solely require presence of functional receptors in membrane of cells destined to die. For example, death mediated by activation of APO/FAS only occurs during certain states of maturation of progenitor cells of T or B–Iymphocytes. In the case of TNF, receptors of this cytokine are present in all cells of the body, but they only activate the death program when transformation toward a tumor–like cell or virar infection produces an internal cellular change that allows the death program to have an effect. In addition, ligands associated to induction of programmed cellular death do not solely induce death. For example, endothelial cells that cover the interior of the vessels have TNF receptors and their activation is an indispensable step for the inflammatory response. In this case, activation of TNF receptors does not promote cellular death, but rather activation of the, endothelium. However, if endothelial cells cultivated in vitro are treated with protein or RNA synthesis inhibitors such as actinomycin D or cycloheximyde, TNF induce death by apoptosis. This observation demostrates that TNF ignites the death program in all cells it stimulates, but cells actively express gene products that protect them; in addition, if these products are not synthesized, the death program follows its course and the endothelial cell dies by apoptosis. The second activation mechanism of the death program consists of a constituent ignition as part of the state of maturation or differentiation characteristic of a cellular lineage. In these cases, death is prevented by action of survival factors, also denominated trophic factors, which if present in the environment interfere with execution of the death program (figure 1, diagram II–b).
Such is the case of neurons dependent on neural growth factor (NGF) or of interleukin–3 (IL–3)–dependent thymocytes. The NGF system participates in neuronal maturation and during selection of functional synapses in the nervous system 5,8,11, whereas the IL–3 system is important during hematopoietic maturation 1–3, 20.

GENETIC CELLULAR DEATH PROGRAM
Experiments with inhibitors of protein and RNA synthesis showed protection of death of IL–3 dependent thymocytes and NGF–dependent neurons. Exposure to these factors promotes survival of a greater number of cells and extends the lifetime of cells with respect to control cultures with no factors.
These were the first indirect evidences of the existence of genes whose expression participates actively in the process of cellular death. Studies on gene expression during death of intercostal muscles of caterpillars of the lepidopter Manduca sexta showed the existence of gene groups specifically expressed during death by apoptosis; complementary to this, gene groups were also found that stopped expressing themselves prior to death of tissues 15. Definitive confirmation of the existence of genes that in time–ordered expression controled the death process carne from studies carried out in the nematode Caenorhabditis elegans 5 , in which only specific cells die at a particular time of development. The majority of cellular deaths in Caenorhabditis elegans is not influenced by neighboring cells or by the position they occupy within these organisms. This suggests that there are no factors released by other cells responsible for activating the death program and that, death is determined genetically by cellular lineage. The study of genetic control of morphogenesis and organogenesis of different larva and adult animal stages of this nematode, resulted in a powerful tool for analysis of genes that participate in the death program. One experimental advantage of this system resides in precise knowledge of the embryonic origin of each cell of the adult animal and which of these somatic cells of this nematode die during development. Thus for example in Caenorhabditis elegans 131 somatic cells die by apoptosis during the last stage of larva development that gives origin to the adult organism 5. Mutagenesis and genetic complementation studies in this organism demonstrated the existence of at least 14 genes that comprise the death programo Some of these compromise the cell–to–death process, while others are program effectors (such as genes ced–3 and ced–4), and still others are required to destroy and dispose of the cellular corpse (such as genes ced–I, ced–2, ced–5, ced–7, ced–B, ced10 and nuc–f). As previously mentioned, genes have been identified (such as ced–9) whose physiologic function consists of interferring with execution of the death program 5,12,17.

ICE AND BCL–2, TWO GENES WITH ANTAGONISTIC FUNCTIONS WITHIN THE DEATH PROGRAM
Although in the nematode Caenorhabditis elegans it was possible to identify several genes that participate in the death program, to date only for genes ced–3 and ced–9 have genes been identified in mammals with homologous sequences and functions (figure 3). Recently, introduction of gene ced–4 to mammals cells revealed the possibility that a gene with a homology function exists, which is essential in activation of proteases during execution phase. The possibility that this protein has a different sequence in nernatodes and mammals would explain why, despite multiple efforts of cloning by homologous hybridization, a gene with homology to gene ced–4 has not been found. In the case of gene ced–3, it was found that in organisms that lack functional alleles of this gene cellular death does not occur; thus gives origin to cells which normally not present in these organisms. Surprisingly, these supernumerary cells, which normally die, turn out to be functional cells that even able to substitute the function of they sisters that norrnally do not die when that eliminated by microsurgery with laser radiation. Cloning and sequence of the gene ced–3 revealed great homology in sequence and later in function with the cysteil–protease ICE, whose function is associated with proteolytic maturation of interleukine–1, from which it derives its name (Interleukin–1 converting enzyme) 21,22. The methodologic possibility of introducing genes of mammals into Caenorhabditis elegans cells (transfection) allowed to introduce the gene that codifies mammal enzyme ICE to genetically deficient nematodes of the gene ced–3 (ced–3–organisms). These experiments demonstrated that enzyme ICE expression can replace the function of the codified product with the gene ced3 (genetic complementation), allowing cellular death by apoptosis to appear in the correct cells at the adequate moment of development. This finding gave way to the cloning of one long list of genes whose sequence is homologous to ICE. These genes codify for endopeptidases denominated caspases, a name they derived from their specificity (cysteil aspartate acid peptidases); these enzymes have been identified in mice and humans 21–22.
Through these transfection studies, it has been determined that expression of caspase 3, 6, 7, 8, and 10 is indispensable for death by apoptosis in many cellular types of mammals. Activation of these proteases occurs in two sequential stages. Caspases 8 and 10 (also denominated FLlCE and Mch4, respectively) participate in the activation phase of the death program and are activated in response to occupation of TNF receptor type 1 or APO/FAS. On the other hand, caspases 3, 6, 7, and 9 (also denominated CPP32, Mch2, IT ICE LAP3, and ICE–LAP6, respectively), comprise the execution phase. These caspases can be activated as the result of mitochondrial damage or by other mitochondrial independent mechanisms not yet characterized (figure 3). Study of the family of caspases demonstrated that the majority cells express these enzymes in the form of inactive or zymogenous precursors that can be activated through proteolysis by members of the same family in a manner analogous to the cascade of proteases that participate in coagulation. Identification of these enzymes explains why the addition protease inhibitor can interfere with death by apoptosis in cultures of neurons without neurotrophic factors or thymocytes without inter–leukine–3 (IL–3) 23.

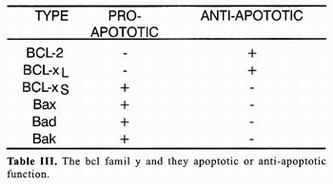
The Caenorhabditis elegans gene ced–9 interrupts the death program, which explains why in organisms that lack functional alleles of this gene (organism ced–9) many more cells die that under normal conditions 3. A portion of the gene ced–9 sequence displays great homology with gene BCL–2, initially identified as a gene associated with development of granulocytic cell B leukemias, hence its name (B–cell–Ieukemia gene 2). Genetic complementation experiments utilizing genetically ced–9–deficient nematodes showed that BCL–2 can replace ced–9 function. These experiments implicitly embrace the concept that genes that control cellular death by apoptosis and the death program itself have been conserved throughout evolution from nematodes, a primitive group of multicellular animals, ta superior vertebrates 24. Table 1 enumerates some BCL–2 homologs present in humans and effects of its expression on the death process. The precise mecha–nism by which BCL–2 interferes with the death program is still unknown despite of an enormous effort to understand' its function. Nevertheless, it has been possible to establish that its protective capacity resides in the possibility of forming BCL 2=BCL–2 homodimers, because versions of BCL–2 devoid of dimerization dominion do not possess antiapoptotic activity. Surpri–singly, over–expression of some members of the family of BCL–2 such as the Bax protein, promotes death. Detailed analysis of Bax protein sequence demons–trated partial homology with BCL–2, since only Bax displays dimerization dominion but lacks a second do–minion present in BCL–2. Bax protein promotes death by interferring with formation of BCL–2=BCL–2 homo–dimers in favor of formation of BCL–2=Bax heterodimers that cannot interfere with death. Comparative study of structures of other members of the family and in vitro protein interaction tests demonstrated that protective or inductive effect of death depends on the possibility of forming furnace or heterodimers among themselves 25. Therefore, the destiny of cellular life or death depends not so much on whether so me BCL–2 family members are express–ed or not, but on balance among members of this family that promote apoptotic death and those that prevent it as is shown in table 1. Among genes with anti–apoptotic activity there are certain viral genes such as crm–A that are codified in the genome of some adenovirus. Peptides codified by these genes belong to the group of serpins, peptides that act as competitive inhibitors of acid cisteilproteases such as caspases. Other serpins have been identified in virus of other species, even in virus of insects such as baculovirus. Ample distribution of these inhibitors such as genes expressed in early stages of virar infection supports the hypothesis that apoptotic death is implied in elimination of cells infected by virus.
OXIDATIVE STRESS, METABOLlSM OF SPHINGOLlPIDS AND ARAQUIDONIC ACID, AND APOPTOTIC DEATH
In addition to activation of the caspase cascade during death by apoptosis, severaI biochemical processes are activated. These have been considered parallel tracts that lead to cellular death. Alterations in the mitochondrial operation have been associated to activation of the cascade of caspases. These include transitory drop in mitochondrial membrane potential, liberation of cytochrome C, and production of free radicals derived from the oxygen. In some cells, these changes appear essential for the cell execution phase. On the one hand, liberation of cytochrome e is an indispensable step in activation of a second series of caspases that in their activation require proteins such as that codified by the Caenorhabditis elegans ced–4 gene. On the other hand, alterations that allow liberation of cytochrome C affect the respiratory chain, permitting an increase in unpaired electron generation and therefore production of free radicals. This increase, mainly of reactive forms of oxygen, creates unspecific oxidative stress that leads to oxidative deterioration of nucleic lipids, proteins and acids, which in itself can lead to cellular death 23.
The importance of oxidative stress can be appraised by the fact that various antioxidant agents intenere with cellular death. Protection conferred by expression of the gene BCL–2 appears to be due to the fact that its gene product inteneres precisely with events of the phase of execution that lead to activation of post–mitochondrial caspases (figure 3). In addition to activation of the cascade of caspases and oxidative stress, other biochemical processes that contribute to the mechanism of cellular death have been identifled. On the one hand is activation of sphingomyelinase that hydrolyzes sphingomyeline producing ceramide, which acts as second messenger able to induce cytostasis (cellular proliferation arrest) or cellular death by apoptosis, depending on the cellular target. On the other hand, activation of A2 type phospholipase (PI–a2) in response to apoptotic stimuli such as TNF produces increase in metabolism of the araquidonic acid that has been associated to cellular death, possibly due to generation of free radicals. Although it is still not known which metabolites of araquidonic acid (prostaglandins, leukotrienes, or thromboxanes) are relevant to death by apoptosis, there are various reports that demonstrate clear correlation between activity of PI–a2 and activation of the death program 22–26.
BIOLOGICAL MEANING OF APOPTOSIS AND PROGRAMMED CELLULAR DEATH
We have already mentioned that programmed cellular death allows us to establish a selection system, eliminating only certain cells within a tissue. On the one hand, by means of a negative selection system, it is possible to eliminate cells potentially dangerous to the organism, as in the case of tumor or cells infected with virus. On the other hand, by means of a positive selection system it is possible to eliminate dysfunctional cells, as in the case of neurons that did not manage to establish functional synapses or elimination of T or B Iymphocytes whose reactivity is directed against its own organism antigens. Additionally is appearance of death of tissue and organs with obsolete functions or reminiscent of the evolutionary past of the organism. An example of the first type of death is reabsorption of tissues and organs during passage from one to another larva stage or metamorphosis of insects and amphibians. An example of the second type of death is represented by reabsorption of Müllerian conduits in males or of mesonephros in females during embryonic development. These types of selections assure that tissues are constituted of cells that perform their function in an optimal manner. Experiments with ced9 organisms of Caenorhabditis elegans show that regulation of the death program constitutes a source of adaptive plasticity, in that cells normally destined to die do not, it is possible that they acquire new functions absent previously. Therefore the population of supernumerary cells constitutes an adaptive reservoir of great value from the evolutionary point of view 27–29.
CONCLUSION
During different stages of development of complex multicellular organisms, cellular death or death and reabsorption of tissues or complete organs, occur(s). It is postulated that this type of cellular death along with cellular proliferation and differentiation form part of the processes that determine size, cellular composition, and function of different organs. This type of cellular death is termed apoptosis or programmed cellular death. It corresponds to a process in which cells that die play an active role in their death and a process that is different from incidental death by hypoxia, poisoning, or trauma (death by necrosis), in which the cell dies victim of circumstances. Nevertheless, in the latter process, the main physiologic difference between necrosis and apoptosis consists of absence of inflammatory response. In view of the fact that cellular death is controlled by genetic information, this process is also subject to the effect of mutations, which along with selective pressures of the environment constitute the raw materials of evolution.
Alterations in regulation of the death process could result in retardation or absence of death, leaving supernumerary cells open to the possibility of forming new cellular interactions, acquiring new functions, and facing new demands. There still remains to be identified genes whose expression control constitutive expression of the cellular death program in Iymphocytes and neurons. On the other hand, there are genes with a protective effect, such as BCL–2 and BCL–XL, for which despite knowledge of genetic products and functional result of their over–expression, biochemical bases of their operation are not yet known in detail. Better understanding of the biochemical mechanisms that lead to death will be very usefulto understand and possibly treat diseases such as Alzheimer's, AIOS and development of tumors that result from alterations in the cellular death program.
ACKNOWLEDGMENTS
This work was partially supported by grants 34823–M from CONACyT, México, PAPIIT, DGAPA–UNAM (IN–211402 and IX–210604), and PAPCA FES–Iztacala, UNAM.
REFERENCES
1. Kerr JFR, Wyllie AH and Currie AR. Apoptosis: a basic biologicalphenomenon with wide ranging implications in tissue kinetics. Sr J Cancer 1972;26:239. [ Links ]
2. Kerr JFR. History of the events leading to the formulation of theapoptosis concept. Toxicology 2002; 181:471-4. [ Links ]
3. Duke RC, Ojicus DM and Young DE. Cell suicide in health and disease. Sci Amer 1996;52:48-55. [ Links ]
4. Dikranian K, Ishimary MJ, TenkovaT, Labruyere J, Qin YQ, Ikonomidou C, Olney JW. Apoptosis in the in vivo mammalian forebrain. Neurobiol Dis 2001;8:359-79. [ Links ]
5. Ellis RE, Yuan I and Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol 1991;7:663-98. [ Links ]
6. Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci 1991;14:453-501. [ Links ]
7. Gould TW, Oppenheim RW. Steppong stone to death. Nat Neurosci 2001;4:1053-4. [ Links ]
8. Allsopp TE , Wyatt S Paterson HF, Davies AM. The protooncogene BCL-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell 1993;73:295-307. [ Links ]
9. Vies DI, Sorenson CM, Shutter IR, Korsmeyer SI. BcI-2-deficient mice demonstrate fulminant Iymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993;75:229-40. [ Links ]
10. Kumar S, Vaux DL. Apoptosis. A cinderella caspase takescenter stage. Science 2002; 297:1290-1. [ Links ]
11. Raff MC. Social controls on cell survival and cel! death. Nature 1992;356:397-400. [ Links ]
12. Vaux DL, Flavell RA. Apoptosis genes and autoimmunity. Curr Opin Immunol 2000;12:719-24. [ Links ]
13. Mirkes PE. Warkany Lecture: to die or not to die, the role ofapoptosis in normal and abnormal mammalian development. Teratology 2002;65:228-39. [ Links ]
14. Peitsch MC, Mannherz HT and Tschopp I. The apoptosis endonucleases: cleaning alter cell death I. Trends Cell Biol 1994;4:37-41. [ Links ]
15. Tibbetts MD, Zheng L, Lenardo MJ. The death effector domain protein family regulators of cellular homeostasis. Nat Immunol 2003; 4:404-9. [ Links ]
16. Roben GM. Caspases: the executioners of apoptosis. Biochem 1997;326:1-16. [ Links ]
17. Golstein P. Controlling cell death. Science 1997;275:1081-2. [ Links ]
18. Feinman R, Koury J, Thames M, Barlogie B, Epstein J, SiegelDS. Role of NF kappaB in the rescuer of multiple myeloma cells from glucocorticoid-induced apoptosis by BCL-2. Blood 1999;93:3044-52. [ Links ]
19. Walton KM, DiRocco R, Bartlett BA, Koury E, Marcy VR, JarvisS, Schaefer EM, Bhat RV. Activation of p53MAPK in microglia after ischemia. J Neurochem 1998; 70:1764-7. [ Links ]
20. Koury MJ, Sawyer ST, Brandt SJ. New insights into erythropoiesis . Curr Opin HematoI 2002;9:93-100. [ Links ]
21. Wong GHW and Goeddel D. Fas antigen and p55 TNF receptorsignal apoptosis through distinct pathways. IlmmunoI 1994;152:17511-755. [ Links ]
22. Koury MI. Programmed cell death (apoptosis) in hematopoiesis. Exp Hematol 1992;20:391-4. [ Links ]
23. Marsden VS, O'Connor L, O'Reilly LA, Silke J, Metcalf D, Ekert PG, Nicholson DW, Vaux DL, Bouillet P Adams JM, Strasser A. Apoptosis initated by BCL-2-regelated caspase activation independently of the cytochrome c/Apat-1/caspase-9 apoptosome. Nature 2002; 419:634-7. [ Links ]
24. Yaginuma H, Sato N, Homma S, Oppenheim RW. Roles of caspases in the programmed cell death of motoneurons in vivo. Arch Histol Cytol 2001; 64:461-74. [ Links ]
25. Kerr DA, Larsen T, Cook SH, Fannjiang YR, Choi E, Griffin DE, et al. BCL-2 and BAX protect adult mice from lethal sindbis virus infection but no protect spinal cord motor neurons or prevent paralysis. J Virol 2002;76:10393-400. [ Links ]
26. Regula KM, Ens K, Kirshenbaum LA. Mitochondria-assisted cellsuicide: a license to kill. J. Mol Cell CardioI 2003;35:559-67. [ Links ]
27. Mattson MP, Liu D. Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem Biophys Res Commun 2003;304:539-549. [ Links ]
28. Borutaite V, Brown GC. Mitochondrial in apoptosis of ischemic heart FEBS Lett 2003;541:1-5. [ Links ]
29. Wong GH, Kaspar RL, Zweiger G, Carlson C, Fong SE, Ehsani N, et al. Strategies for manipulating apoptosis for cancer hetrapy with tumor necrosis factor and Iymphotoxin. J Cell Biochem 1996;60:56-60. [ Links ]

