











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Peutz-Jeghers (SPJ) es una entidad poco frecuente, de herencia autosómica dominante, con expresividad variable y penetrancia completa. En un alto porcentaje es causado por mutaciones del gen STK11, localizado en el brazo corto del cromosoma 19, región 1, banda 3, subanda 3. En 1921, el doctor Johannes Peutz describe varios miembros de una familia con pigmentación mucocutánea y poliposis intestinal; posteriormente en 1949, Jeghers reconoce su carácter hereditario y su asociación con cáncer. Fue hasta 1954, cuando el doctor Bruwer acuñó el término de SPJ.1
Esta enfermedad está caracterizada por la presencia de pigmentaciones puntiformes en la mucosa oral, labios, región palmar y plantar y, en ocasiones en la región perianal, asociada con poliposis múltiple gastrointestinal.2 Para su diagnóstico, la Organización Mundial de la Salud exige cuatro criterios: 1) la presencia de tres o más pólipos de Peutz-Jeghers confirmados histológicamente; 2) cualquier número de pólipos de Peutz-Jeghers con historia familiar de este síndrome; 3) pigmentación mucocutánea característica, con historia familiar de SPJ, y 4) cualquier número de pólipos de Peutz-Jeghers y pigmentación mucocutánea característica.3
En Estados Unidos de Norteamérica se ha descrito que ocurre un caso por cada 8,300 nacimientos. En el servicio de Anatomía Patológica de nuestro hospital se reportan alrededor de tres nuevos casos al año en edades pediátricas. Esta entidad se caracteriza por poliposis hamartomatosa, que es una proliferación celular benigna, y que se puede localizar a lo largo de todo el tubo digestivo; las porciones más afectadas son el yeyuno-íleon, apareciendo en colon y recto en 33% y en estómago en 25%. El cólico abdominal recurrente, que es causado por la intususcepción transitoria de los pólipos, suele ser el síntoma más frecuente, mientras que la hemorragia es menos frecuente. También se han descrito tumores malignos del aparato gastrointestinal.4
A los pacientes con SPJ se brinda tratamiento sintomático en los eventos agudos. Se recomienda el seguimiento periódico para detectar las lesiones tumorales tempranas y realización de polipectomías por endoscopía en pólipos grandes con el fin de evitar la transformación a un hamartomatoma-adenomacarcinoma
Por su parte, la enfermedad de Wilson (EW) es un trastorno del metabolismo del cobre que se hereda en forma autosómica recesiva. Se ha reportado una tasa de portadores de uno por cada 90 casos.5 El gen ATP7B se localiza en el brazo largo del cromosoma 13, región 1, banda 4, subanda 3, que codifica para la proteína ATPasa tipo P transportadora de cobre (ATPasa de Wilson). Desde su descubrimiento se han descrito más de 800 mutaciones del gen ATP7B, siendo la más común c.3207C>A (p.His1069Gln).
La patogénesis de la enfermedad de Wilson se debe a una alteración del almacenamiento y movilización del cobre de los lisosomas hepatocelulares para su excreción biliar. La acumulación excesiva de cobre en los diferentes tejidos es responsable tanto de la disfunción hepática como de los trastornos neurológicos y de la formación de los anillos de Kayser-Fleischer. Estos últimos sugieren fuertemente esta enfermedad. Los signos neurológicos se manifiestan por lo general entre la segunda y tercera década de la vida, y se caracterizan por Parkinson, ataxia y síndrome distónico. El anillo de Kayser-Fleischer puede estar presente sólo en 40% de los pacientes.6
Se considera que en la EW resulta de un defecto en la síntesis de ceruloplasmina o de la presencia de una proteína anormal con gran afinidad por el cobre, que impide la unión de este catión a la apoceruloplasmina. Así, la mayoría de los pacientes tienen niveles de ceruloplasmina < 20 mg/dL; nivel plasmático de cobre total < 60 μg/dL y cobre en orina de 24 horas > 100 μg, mientras que el cobre hepático > 250 μg por gramo de tejido. Dentro del tratamiento, existen dos enfoques: quelantes (penicilamina o trientina) o prevención de la absorción mediante la ingesta de altas concentraciones de zinc.7
En este artículo describimos (hasta nuestro conocimiento) el primer caso de la presentación simultánea de enfermedad de Wilson en una paciente con síndrome de Peutz-Jegher.
Presentación del caso
Se trata de paciente del sexo femenino, con antecedente de abuelo materno con SPJ. A los siete años presentó anemia severa persistente a pesar del tratamiento y debido a sus antecedentes familiares, en un hospital de segundo nivel, se realizó endoscopía y por reporte histopatológico de pólipos hamartomatosos sin displasia se llegó al diagnóstico de síndrome de Peutz-Jeghers. Desde entonces se ha vigilado realizando endoscopías de manera periódica, así como polipectomías.
Aproximadamente un año después se detectó elevación de enzimas hepáticas a más de dos veces su valor normal, iniciándose abordaje diagnóstico; sin embargo, tras más de tres años y al no presentar mejoría ni identificar la causa fue referida al Centro Médico 20 de Noviembre del ISSSTE.
A su ingreso, la paciente se encontró alerta y orientada. En la exploración de fondo de ojo no se identificaron anillos de Kayser-Fleischer. Se observaron lesiones pigmentadas en el labio inferior; el resto fue normal, incluyendo la exploración neurológica y sin ninguna manifestación psiquiátrica.
Los estudios de laboratorio mostraron los siguientes datos: ALT 314, AST: 268, GGT: 784, FA: 860 BT: 0.36. La serología para hepatitis B, C y VIH resultó negativa, lo mismo ocurrió en la detección de anticuerpos para enfermedades autoinmunes.
El ultrasonido hepático mostró el hígado de forma y situación habitual, con bordes regulares y bien definidos. El parénquima fue de ecogenicidad aumentada de manera difusa +. No se observó dilatación de la vía biliar intrahepática o extrahepática.
La tomografía axial computarizada (TAC) evidenció la glándula hepática con situación y morfología normales, con bordes regulares y bien definidos, parénquima homogéneo y de densidad conservada, mientras que en la resonancia magnética (RMN) de cráneo no se detectaron alteraciones.
Por colangiorresonancia no hubo evidencia de lesión en parénquima hepático; las vías biliares intrahepáticas se mostraron normales y el colédoco intrapancreático conservado, sin apreciar obstrucción.
Una vez que se descartaron enfermedades infecciosas y autoinmunes, se decidió realizar biopsia de hígado. En la biopsia hepática se reportó la presencia de puentes de tejido fibroso, mínima proliferación de conductos biliares, inflamación leve del predominio mononuclear y plasmocitosis. Los hepatocitos tuvieron degeneración globosa y escasos cuerpos de Mallory-Denk, de localización periportal y gránulos de proteína fijada al cobre que se tiñó de café oscuro con la técnica de orceína acética de Shikata. Algunos hepatocitos mostraron acidofilia citoplásmica y cuerpos apoptósicos; no hubo glucogenización nuclear ni esteatosis (Figura 1). Por lo anterior, se estableció que los hallazgos eran compatibles con enfermedad de Wilson en estadio precirrótico.
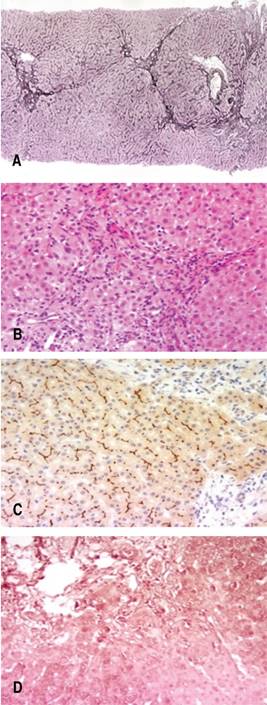
Figura 1: A) Reticulina. Parénquima hepático que muestra patrón macronodular. B) Hematoxilina eosina. Infiltrado inflamatorio mixto, aumento en el número de conductos biliares, acúmulo de pigmento biliar, pseudorrosetas. C) BSEP (Bile Salt Export Pump) expresado a lo largo de los márgenes canaliculares. D) Orceína que muestra depósito de proteína ligada al cobre.
Posterior al reporte de patología se hicieron otros estudios. Así, la ceruloplasmina sérica: 3 mg/dL (normal: 20-60 mg/dL); cobre urinario: 1,050 μg/dL (normal: 15-60); cobre sérico: 2,043 UG/L (normal: 700-1,750), mientras que la determinación de cobre hepático fue de 80 μg/g (normal: 10-35).
Con estos últimos resultados se concluyó que se trataba de enfermedad de Wilson con afección hepática, por lo que se inició el tratamiento con D-penicilamina a dosis de 20 mg/kg por día. En los controles de pruebas funcionales hepáticas tres meses posteriores al tratamiento se observó mejoría: AST de 131, ALT de 160, fosfatasa alcalina de 664, albúmina de 4.36, colesterol de 158, BT de 2.14, BD de 0.24 y ceruloplasmina sérica de 32.6 mg/dL.
Discusión
Después de revisar de manera intencionada la bibliografía y, hasta nuestro conocimiento, creemos que este es el primer reporte de un caso donde coexisten el SPJ con la EW, las cuales son dos entidades que tienen un componente genético no relacionado entre sí.
Con respecto a la EW, la elevación de las transaminasas como único hallazgo inicial se considera un reto para el diagnóstico.8 Ante un niño asintomático con elevación de transaminasas de manera crónica y sin síntomas neurológicos se deben descartar patologías como problemas infecciosos, tóxicos, autoinmunes, hemocromatosis, enfermedad celiaca, miopatías o deficiencia de alfa-1 antitripsina,9 lo cual se llevó a cabo en nuestra paciente.
La EW se caracteriza por incapacidad para excretar cobre en la bilis secundaria a la deficiencia en la proteína transportadora ATP7B. La actividad de las transaminasas se observa aumentada entre 18 y 23% de los pacientes; siendo más común de dos a cinco veces su valor como fue en el presente caso.10 Habitualmente, la enfermedad hepática puede preceder a las manifestaciones neurológicas hasta por 10 años. Dentro de estas últimas, podemos mencionar temblores, anomalías del habla, distonía, convulsiones y signos cerebelosos.11 Además, puede existir hemolítica hasta en 12% de los pacientes.12
Los anillos de Kayser-Fleischer se han descrito como un sello distintivo, y su densidad se correlaciona con la gravedad de la enfermedad. Sin embargo, en pacientes que sólo tienen afección hepática, hasta 50% carecen de anillos, tal como sucedió en el caso que nos ocupa. Se ha reportado que están presentes en la gran mayoría de los pacientes con síntomas neurológicos, y más aún con síntomas psiquiátricos (98%). Además, se debe considerar que estos anillos no son patognomónicos, ya que se encuentran en la cirrosis criptogénica o en hepatitis crónica.13
El cobre extremadamente bajo (< 5 mg/dL) deberá considerarse como un criterio de sospecha para diagnóstico de EW, en especial en asociación con anillos de Kayser-Fleischer. No obstante, una concentración dentro de rangos normales no excluye el diagnóstico, esto es debido a la presencia de inflamación, puesto que el cobre es un reactante de fase aguda o por efecto de los estrógenos. Concentraciones de cobre sérico total normales o elevadas, junto con la disminución de los niveles de ceruloplasmina sérica, indican un incremento de la concentración de cobre “libre”. En individuos sanos, esta concentración representa menos de 10% de la concentración de cobre sérico total, mientras que en la EW este porcentaje puede elevarse hasta 30-50%. Así, en la mayoría de pacientes no tratados se observan concentraciones de cobre no unido a ceruloplasmina superiores a 3.15 μmol/L (intervalo de referencia: 0.79-1.57 μmol/L, o bien, 5-10 μg/dL]).14
La medición cuantitativa de cobre hepático mediante biopsia se considera la prueba diagnóstica definitiva para EW, lo cual se reserva para casos en los que otras pruebas no muestran resultados definitivos. Tiene una sensibilidad de 83.3 a 96.5% y una especificidad de 95.4%, pero su distribución en el parénquima no es uniforme. En nuestra paciente, el cobre en tejido hepático fue de 80 μg/g; sin embargo, el valor diagnóstico debe ser > 200 μg/g. Se conoce que esta cifra sólo es alcanzada en 83% de los enfermos; cifras menores se pueden encontrar porque la biopsia incluye nódulos de regeneración en los que el contenido de cobre es prácticamente nulo. Se requiere, por tanto, un adecuado tamaño de muestra (1 cm × 1.6 mm), el cual se seca en un recipiente exento de elementos a 90 oC hasta conseguir una masa constante y posteriormente se somete a hidrólisis ácida con HNO3 ultrapuro.14,15 También se ha descrito que los pacientes heterocigotos pueden presentar menor cantidad de cobre hepático, entre 50 y 250 μg/g tejido.
También para el diagnóstico de la EW se ha propuesto un sistema de puntuación desarrollado en Leipzig en 2001, el cual toma como variables los anillos de Kayser-Fleischer, síntomas neurológicos, ceruloplasminemia, anemia hemolítica, cuantificación de cobre hepático, cupruria y análisis de mutaciones. Una puntuación > 4 establece el diagnóstico.16 Si nos basamos en este sistema de puntuación, a nuestra paciente le corresponderán tres puntos basados en la cupruria, cobre sérico y hepático.
Si bien la paciente que reportamos mostró disminución de los niveles de transaminasas posterior a tres meses de tratamiento, está documentado que el funcionamiento hepático puede volver a la normalidad hasta después de dos años de tratamiento en pacientes con y sin cirrosis, sin progresión del daño hepático.
Por último, es necesario comentar que la confirmación molecular de ambas entidades podrá fundamentar la asociación de SPJ y EW en una misma paciente; sin embargo, por el momento no contamos con este recurso en la institución.

