










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de investigación clínica
versión On-line ISSN 2564-8896versión impresa ISSN 0034-8376
Rev. invest. clín. vol.58 no.4 Ciudad de México jul./ago. 2006
Artículo de revisión
Hipotiroidismo neonatal: fisiopatogenia, aspectos moleculares, metabólicos y clínicos
Neonatal hypothyroidism. Pathophysiogenic, molecular and clinical aspects
Juan Carlos Solis,* Carlos Valverde**
* Division of Endocrinology, Metabolism, and Molecular Medicine, Feinberg School of Medicine, Northwestern University, Chicago.
** Laboratorio de Fisiología Evolutiva, Departamento de Neurobiología Celular y Molecular. Instituto de Neurobiología, Universidad Nacional Autónoma de México; Campus UNAM–UAQ.
Reimpresos:
Dr. Juan Carlos Solis
Division of Endocrinology, Metabolism, and Molecular Medicine
Department of Medicine, Feinberg School of
Medicine, Northwestern University,
Tarry Building 15–710, 303 E Chicago Avenue,
Chicago IL, 60611; USA
Tel: +312.503.4131,
Correo electrónico: j–solis@northwestern.edu
Recibido el 9 de julio de 2004.
Aceptado el 1 de marzo de 2006.
ABSTRACT
This review provides an updated summary on both the clinical and diagnostic aspects of neonatal hypothyroidism (NeH); as well as on the molecular and pathophysiologic processes known to be involved in the installment of this important hormonal deficiency. Current information regarding its etiology and pathogenesis has allowed classigying NeH in three major groups: endemic, transient, and sporadic hypothyroidism. The later corresponds to congenital hypothyroidism and encompasses a broad spectrum of hereditary disorders causing hypothyroidism in newborns and young children. These congenital disorders include hypothalamic–pituitary or thyroid dysgenesis and/or dyshormonogenesis, as well as hyporesponsiveness or resistance to either TRH, TSH or to thyroid hormones. The introduction of national screening programs for NeH have overcome the difficulties in the early diagnosis thus helping to prevent its serious and irreversible consequences on intellectual and physical development. Concomitantly, an increase in the need for complementary etiologic and molecular diagnosis has risen. The current capability to perform a fine and precise diagnose is crucial both for treatment of the affected infant and for genetic counseling of the family. Although incomplete, available epidemiological information in Mexico indicates that NeH prevalence can be as high as twice that in other developed world countries. On these bases, national public health policies and epidemiological surveyance must be strengthen not only to identify, diagnose and timely treat, but to prevent and eradicate endemic NeH.
Key words. Neonatal hypothyroidism. Iodine. Thyroid hormones. Thyroid gland. Congenital hypothyroidism.
RESUMEN
Este trabajo revisa algunos aspectos del conocimiento actual sobre la fisiopatogenia, los hallazgos clínicos y el diagnóstico bioquímico y molecular del hipotiroidismo neonatal (HNe). El término HNe denota un conjunto de entidades clínicamente pleomórficas, que invariablemente cursan con una disminución en el aporte; o bien, en la disponibilidad celular y/o en la respuesta a las hormonas tiroideas (HT) durante la etapa perinatal. Las HT o yodotironinas son indispensables para la morfogénesis y maduración funcional normal de prácticamente todos los tejidos en el organismo, y su participación es crucial en el caso del sistema nervioso. La información actual permite realizar una clasificación del HNe tanto en términos etio y fisiopatogénicos, como en el contexto del substrato genético que los determina. Así, se reconocen tres grandes tipos de HNe: el endémico, el transitorio y el esporádico. Este último grupo de HNe incluye los defectos hipotálamo–hipofisiarios, los trastornos ontogenéticos o disgenesias tiroideas, la resistencia periférica a las HT y las dishormonogénesis. Por otra parte, en la comunidad internacional existe una creciente preocupación por la contaminación ambiental debida a órgano–halógenos antropogénicos. Estos compuestos han mostrado su potencial como agentes distiroideos en animales de experimentación y en algunos estudios clínicos. En México, tanto la distribución geográfica y prevalencia del HNe, como la deficiencia de yodo y otros micronutrimentos en la dieta, se han analizado de manera esporádica y no sistemática. Aunque incompleta, la información disponible sugiere que en nuestro país la prevalencia de HNe es sensiblemente mayor que la reportada mundialmente. Contar con información completa y confiable acerca de estos aspectos no es trivial, puesto que su conocimiento permitirá establecer políticas razonadas de salud pública para identificar, diagnosticar y tratar oportunamente el padecimiento; así como para prevenir y erradicar el HNe endémico.
Palabras clave. Yodo. Tiroides. Yodotironinas. Hipotiroidismo neonatal. Hipotiroidismo esporádico congénito.
INTRODUCCIÓN
Las yodotironinas u hormonas tiroideas (HT) controlan de manera directa; o bien, coparticipan en la operación de prácticamente todos los procesos fisiológicos del organismo (Cuadro 1).1–7 Así, en todos los vertebrados durante los llamados periodos críticos del desarrollo, las HT intervienen en la maduración y diferenciación funcional de varios órganos. Tal es el caso del sistema nervioso central en desarrollo, en el que un aporte insuficiente de HT en tiempo y lugar, resulta en anomalías permanentes.8,9 El objetivo de este trabajo es revisar los avances recientes sobre la fisiopatogenia, hallazgos clínicos y el diagnóstico bioquímico y molecular del hipotiroidismo neonatal (HNe). A pesar de las medidas adoptadas para su erradicación, los trastornos secundarios a deficiencia de yodo continúan siendo un grave problema de salud pública mundial. Por otra parte, y no obstante su menor incidencia, también es importante reconocer y diagnosticar oportunamente los trastornos glandulares y metabólicos que conducen al HNe. Las pruebas bioquímicas de tamizaje neonatal y las técnicas modernas de imagen en medicina nuclear, han facilitado la detección temprana del HNe. Sin embargo, estas pruebas requieren complementarse con la identificación etiológica y/o genética del padecimiento. Esta precisión diagnóstica es fundamental tanto para el tratamiento adecuado del probando como para el consejo genético de su familia.
IMPORTANCIA BIOLÓGICA DEL YODO
Se considera que un elemento químico es esencial para el organismo cuando su disponibilidad por abajo de cierto límite, invariablemente provoca disfunción, enfermedad e inclusive la muerte; o bien, cuando ese elemento forma parte de una estructura orgánica.10,11 Se sabe que aproximadamente una tercera parte de los 92 elementos naturales son esenciales para todos los sistemas vivos. Los más abundantes son el hidrógeno, el oxígeno, el carbono y el nitrógeno. Estos cuatro bioelementos tienen números atómicos menores a nueve y representan alrededor de 99% del total de átomos presentes en los seres vivos. Por su abundancia relativa y sus requerimientos dietéticos, los bioelementos restantes se agrupan en macrominerales, elementos traza y elementos ultratraza o micronutrimentos. Los requerimientos de los elementos ultratraza se ubican generalmente en el intervalo de los µg/día y, como su nombre lo indica, son elementos que por su exigua concentración apenas contribuyen con menos de 0.01% del total de átomos en los sistemas vivos.11,12 El bioelemento ultratraza más pesado y rico en electrones es el yodo (I127), un halógeno que pertenece al grupo VILA de la tabla periódica. Solamente tres de los halógenos naturales son esenciales para la vida: el cloro (Cl2), el bromo (Br2), y el yodo (I2); el flúor (F2) se considera un elemento benéfico, pero no esencial. La abundancia relativa de los halógenos naturales en el agua de mar y en la litosfera (Cuadro 2), contrasta con la notable capacidad del hombre y los cordados en general, así como la de algunas algas del orden de las laminariales, para acumular y concentrar Br2 e I2–13–15 El caso del yodo es especialmente singular puesto que su abundancia en uno y otro medio es 1.000,000 veces menor que la del resto de los halógenos. Sin embargo, y no obstante su escasez, en términos evolutivos el yodo fue seleccionado como la materia prima esencial para la síntesis de HT.15
La captura de halógenos y su incorporación a moléculas biológicas (organificación) es una función omnipresente en la biosfera, y a la fecha se conocen más de 3,800 organohalógenos naturales.16 Sin embargo, para el caso específico del yodo, es importante enfatizar que solamente los vertebrados poseen un órgano especializado en capturar activamente este halógeno y organificarlo en los residuos de tirosina contenidos en la tiroglobulina (Tg), paso crítico en la síntesis de yodotironinas.15 En todos los vertebrados estudiados a la fecha, el halógeno ingresa al organismo a través de los nutrimentos y el agua, y lo hace en la forma de yodo (I2); o bien, como el ion yoduro (I). El I – es rápidamente absorbido en el tracto gastrointestinal y distribuido a los líquidos extracelulares, de donde es ávidamente captado por la glándula tiroides y/o depurado por el riñon.17
LA GLÁNDULA TIROIDES Y LAS YODOTIRONINAS
Entre las glándulas exclusivamente endocrinas o de secreción interna, la tiroides es una glándula singular. Además de ser la más grande, es la única que captura y almacena, en el espacio extracelular, cantidades importantes de yodo y de sus productos de secreción: las yodotironinas. La característica distintiva tanto de los precursores mono– o diyodados (MIT y DIT, respectivamente) como de las HT es la presencia de átomos de yodo en su molécula (Figura 1). Así, por ejemplo, los átomos del halógeno aportan 65% y 58% de las respectivas masas moleculares de la tiroxina o tetrayodotironina (T4) y de la triyodotironina (T3). Aunque la forma de la glándula varía en las diferentes clases de vertebrados, en todos la unidad funcional básica es el folículo tiroideo. Este módulo morfofuncional de la tiroides es una estructura esférica que consta de dos partes:
1. Un epitelio simple de células altamente especializadas denominadas tirocitos o células foliculares.
2. La cavidad folicular, un espacio sacular extracelular central que almacena al coloide tiroideo.
Dependiendo del estado funcional de la glándula y del aporte de yodo en la dieta, el coloide tiroideo es el mayor depósito en el organismo de T4 y T3.15,18
Desarrollo normal de la glándula tiroides
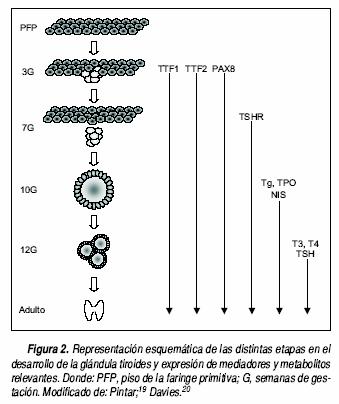
La glándula tiroides es el primer tejido endocrino glandular que aparece en el desarrollo embrionario del ser humano. Como se muestra en la figura 2, en su desarrollo participan diversos factores de transcripción como el TTF1, TTF2 y PAX8 (para una mayor descripción de estos factores véase sección de "Disgenesias Tiroideas")19,20 La glándula se origina a partir de un primordio celular endodérmico localizado en la línea media en el piso de la faringe primitiva, entre el primero y segundo arcos branquiales. Este primordio se invagina y da lugar al divertículo tiroideo el cual, conforme el embrión y la lengua crecen, desciende a través del cuello. A medida que el divertículo desciende se mantiene comunicado con la base de la lengua mediante un tubo estrecho, el conducto tirogloso, cuya abertura lingual se denomina agujero ciego. Posteriormente el divertículo tiroideo se fusiona con el cuerpo ultimobranquial (derivado de la cuarta y quinta bolsas faríngeas), estructura que aporta los precursores de las células parafoliculares, también conocidas como células tipo C, productoras de calcitonina. A las siete semanas de gestación (7G), la glándula tiroides alcanza su ubicación definitiva en la parte anterior del cuello. Este desplazamiento caudal se acompaña por una rápida elongación del conducto tirogloso, el cual eventualmente se fragmenta y degenera. El rudimento tiroideo comienza después a expandirse de forma lateral, lo cual conduce a la formación de su característica estructura bilobulada. La captación de yodo y las primeras evidencias de síntesis de T4 ocurren entre la semana 10–12G. Por otra parte, la hormona estimulante de la tiroides (TSH) fetal solamente es detectable a partir de la semana 12G, aumentando progresivamente durante los meses restantes de la gestación, a la vez que ocurre un incremento en la síntesis y secreción de HT.21–23
Biosíntesis de las hormonas tiroideas
La vía metabólica del yodo que conduce a la biosíntesis de las HT comprende tres pasos secuenciales:
1. El transporte activo (captura) del yoduro al interior de los tirocitos.
2. La oxidación del yoduro.
3. La organifición específica en residuos tirosilo "hormonogénicos" presentes en la molécula de tiroglobulina.17,24
En este proceso de hormonogénesis participan al menos siete diferentes tipos de proteínas:
1. El transportador de yodo (simportador Na/I o NIS).
2. La pendrina (antiportador Cl/I o PDS).
3. La tiroglobulina (Tg).
4. Diferentes chaperonas moleculares (calnexina, BiP).
5. La tiroperoxidasa (TPO).
6. La oxidasa tiroidea de NADPH (THOX).
7. La deshalogenasa tiroidea (tDh).
Tal y como se ilustra en la figura 3, la biosíntesis de las yodotironinas se puede resumir en 10 pasos cruciales.18,24–29
HIPOTIROIDISMO NEONATAL (HNe)
Hallazgos clínicos
Como se resume en el cuadro 1, las HT participan en la regulación; o bien, son esenciales para la operación y función normal de prácticamente todos los procesos celulares del organismo. Estas acciones son principalmente mediadas por su unión a receptores nucleares específicos.18 Como consecuencia de esta diversidad funcional, el cuadro clínico secundario a la deficiencia de HT es pleomórfico y por lo tanto poco específico. Es importante destacar que además de su papel morfogenético durante periodos críticos especie–específicos, la deficiencia de HT disminuye la síntesis y secreción de prácticamente todas las hormonas del organismo, así como de diferentes proteínas estructurales y catalíticas responsables de la homeostasis celular.30–33
Los signos clínicos del paciente con HNe varían de acuerdo con la causa, severidad y duración de la deficiencia hormonal (Cuadro 3).23,34–36 El diagnóstico clínico del HNe puede en ocasiones hacerse al nacimiento, sobre todo cuando se trata de neonatos con agenesia de la glándula tiroides, o con un déficit completo en la síntesis de HT; i.e., cuando la deficiencia in útero no pudo ser compensada por el aporte materno. El diagnóstico también es relativamente sencillo en aquellos neonatos expuestos durante la gestación a cantidades importantes de anticuerpos maternos bloqueadores del receptor a TSH. Sin embargo, el diagnóstico clínico es la excepción. En efecto, se calcula que solamente 5% de los neonatos con HNe pueden ser detectados clínicamente pues por lo general, los signos típicos aparecen después de varias semanas de vida.34,37,38

Por esta razón, es crucial reducir al máximo la ventana de tiempo entre la sospecha diagnóstica y la instauración del tratamiento. Las consecuencias de hacerlo tardíamente pueden ser devastadoras para el desarrollo físico y mental del neonato y tienen un elevado costo familiar y social.8,9,39,40
Tamizaje neonatal
Las consecuencias de la instauración tardía en el tratamiento hacen evidente la enorme importancia del tamizaje neonatal como el recurso diagnóstico imprescindible para el HNe. A este respecto, es importante señalar que en los países desarrollados este recurso diagnóstico se ha implantado de forma extensa desde la década de los 70's.41 En contraste, y no obstante su obligatoriedad desde 1988,42 en México la cobertura nacional del tamizaje neonatal dista de ser completa. El empleo de este recurso diagnóstico en nuestro país consiste en obtener sangre del talón o del cordón umbilical del neonato, la cual se deposita en una tarjeta de papel filtro especial (tarjeta de Guthrie) para la cuantificación posterior de TSH.
En términos generales, el valor de esta hormona se considera sospechoso de hipotiroidismo congénito cuando excede las 10 mUI/mL en sangre de talón, o bien las 15 mUI/mL en muestra de cordón umbilical.43 Una vez confirmado el diagnóstico mediante un perfil tiroideo sérico (TSH, T4 y Tg), se debe proceder a la instauración del tratamiento.
Clasificación del HNe
En la práctica clínica, y tomando en cuenta el sitio anatómico en el cual reside el déficit funcional, tradicionalmente el síndrome de hipotiroidismo se ha clasificado como: primario (déficit tiroideo); secundario (déficit hipofisiario); terciario o central (déficit hipotalámico), y resistencia periférica a la acción de las HT.18 En años recientes diferentes estudios tanto clínicos como experimentales han revelado que el desarrollo de la glándula tiroides, así como la síntesis, almacenamiento y secreción de las HT, dependen de una secuencia precisa y ordenada de eventos bioquímicos y factores de trascripción puntualmente regulados. Así, en la actualidad se considera que la alteración de cualquiera de estos factores o de sus reacciones y procesos intermediarios, brinda las bases para establecer una clasificación etiológica y fisiopatogénica del HNe. Como se resume en el cuadro 4, por el momento esta clasificación incluye tres grandes grupos:

1. El hipotiroidismo endémico causado por la deficiencia en la ingesta de yodo.
2. El hipotiroidismo transitorio.
3. El hipotiroidismo esporádico congénito propiamente dicho.34,38,44
Dentro de este último grupo se incluyen las alteraciones hipotálamo–hipofisiarias; los trastornos ontogenéticos mejor conocidos como disgenesias tiroideas; las alteraciones que conducen a una disminución en la respuesta celular a las HT, y los defectos metabólicos en la síntesis de las HT también conocidos como defectos dishormonogénicos. Para los propósitos de esta revisión resulta más conveniente utilizar esta clasificación etiopatogénica.
Hipotiroidismo endémico
Además de la presencia de una glándula tiroides anatómica y funcionalmente normal, la síntesis de HT depende de manera absoluta del aporte de yodo en la dieta. La carencia de este halógeno provoca los llamados trastornos secundarios a la deficiencia de yodo o IDD.45 Las repercusiones orgánicas y funcionales de esta insuficiencia varían según el estadio ontogenético del individuo (Cuadro 5).46–49 A la fecha, se reconoce que los IDD son la causa más frecuente de daño cerebral y retardo mental prevenible en el mundo. Las evaluaciones más recientes de la Organización Mundial de la Salud indican que aproximadamente 2.2 billones de personas (38% de la población mundial) viven en áreas de bocio endémico; además, se estima que 740 millones padecen IDD, y que de éstos 11.2 millones presentan retraso psicomotor irreversible (cretinismo).50,51 La causa más frecuente de esta deficiencia mental es el hipotiroidismo neonatal. En países desarrollados en donde el aporte dietético de yodo es suficiente, la prevalencia del HNe oscila entre 1/3,000 a 1/ 4,000 recién nacidos vivos.46,52 En los países en vías de desarrollo estas cifras son sensiblemente mayores. En México, en la población que nace en unidades asistenciales de la Secretaría de Salud de todo el país, la prevalencia de HNe oscila entre 1/1,338 a 1/2.572.43,53,54 Aunado a las bajas concentraciones de yodo presentes en áreas bociógenas endémicas, la presencia de agentes bociógenos naturales y la deficiencia de selenio parecen agravar las manifestaciones clínicas del hipotiroidismo.47,55 En nuestro país la información al respecto es escasa y fragmentada.43,56,57
Hipotiroidismo transitorio (HiT)
El HiT se define como aquel detectado al tiempo del tamizaje neonatal que desaparece de manera espontánea y completa con el paso del tiempo. Este tipo de hipotiroidismo es más frecuente en neonatos prematuros y tiene una relación directa y creciente con el grado de prematurez. Dos por ciento de todos los casos de HNe corresponde a neonatos hijos de madres con tiroiditis autoinmune crónica. En estos casos el HiT es causado por la transferencia placentaria de anticuerpos maternos que bloquean el receptor a TSH. Estos anticuerpos maternos gradualmente desaparecen, al igual que el estado de hipotiroidismo. Otra causa de HiT es la exposición de un feto o neonato a preparaciones con yodo orgánico e inorgánico que son comúnmente utilizadas en forma de antisépticos.38,58
En ocasiones el diagnóstico diferencial de HiT puede ser difícil de realizar, e incluso algunos de estos neonatos pueden cursar con niveles elevados de TSH por varios meses. Estos niños deben tratarse al igual que los otros tipos de HNe, hasta que sea evidente que no se requiere reajustar el tratamiento para mantener la tasa de crecimiento, o bien hasta que el niño cumpla cuatro años de edad. Se considera que en este momento se puede optar por suspender el tratamiento por varias semanas para evaluar la función tiroidea y determinar si el infante continúa siendo hipotiroideo.34 Más recientemente, se ha propuesto el uso de TSH recombinante humana, ecosonografía y gammagrafía para distinguir entre las formas transitorias y las permanentes del HNe, evitando así interrumpir el tratamiento del paciente.59
Hipotiroidismo esporádico
Tal y como se ilustra en el cuadro 6, la información actual permite establecer una correlación entre las entidades causales del hipotiroidismo esporádico con el defecto molecular que las origina. De esta forma, se distinguen los siguientes cuatro tipos de hipotiroidismo esporádico:60–62
Defectos hipotálamo–hipofisiarios (DHH)
Por definición, este tipo de HNe implica la falta de estimulación de la glándula tiroides por parte de la TSH. A su vez, esta falla puede deberse a la disfunción en uno o en ambos elementos de la unidad hipotálamo–hipofisiaria. Este tipo de HNe es raro y ocurre en aproximadamente uno de cada 20,000 neonatos. La deficiencia puede obedecer al desarrollo anormal de los componentes del sistema neuroendocrino implicados en regular la función de la glándula tiroides; o bien, a una alteración en la estructura química de la TSH o en los genes reguladores de esta hormona. Actualmente se conoce que en la ontogenia de los tirotropos (células secretoras de TSH) participan al menos cuatro factores de transcripción: HESX1, LHX3, POU1F1 y PROP1. Entre estos defectos las mutaciones reportadas en PROP1 han sido por mucho las más comunes. En murinos, se han identificado defectos en otros factores hipofisiarios, tales como PITX1, PITX2 y GATA2; sin embargo, éstos no han sido reportados en humanos.44,60,63
Es frecuente que el HNe secundario a DHH se asocie a otras deficiencias hormonales hipofisiarias, así como a malformaciones de algunas estructuras cerebrales, tales como el nervio óptico y la médula espinal cervical. Las deficiencias hormonales asociadas, especialmente la deficiencia de cortisol, pueden explicar la alta morbimortalidad que caracteriza a este tipo de HNe. El DHH que no implica disgenesias está relacionado con defectos tanto en el gen de la subunidad beta de la TSH como en el del receptor a TRH. A la fecha se han descrito siete y tres mutaciones distintas, para uno y otro de estos genes.44,60,63–65
Disgenesia tiroidea (DisT)
A la fecha, se considera que entre 80–90% de los casos de HNe son consecuencia de alteraciones en la organogénesis de la glándula tiroides. Estas alteraciones son debidas a mutaciones en los genes involucrados en la diferenciación de los tirocitos; sin embargo, en muchos casos la alteración genética causal no se conoce. Las DisT son primordialmente esporádicas, aunque se han catalogado como una alteración familiar en aproximadamente 2% de los pacientes. Por razones desconocidas, la frecuencia de estas alteraciones es sensiblemente mayor en mujeres que en hombres (2–3:1). Las DisT pueden subdividirse en tres grandes grupos:
a) Tiroides ectópica, usualmente pequeña y sublingual (30–45%).
b) Agenesia (35–40%).
c) Hipoplasia (5% de los casos).23,44,66
Los fenotipos observados en las DisT indican que el mecanismo patogénico puede explicarse por alteraciones en la organogénesis. Hasta el momento se han identificado tres factores de transcripción importantes relacionados con la DisT, los cuales se expresan desde etapas tempranas de la morfogénesis hasta la etapa adulta:21,61,65–70
1. TTF1 (también conocido como NKX2.1). Este factor de transcripción se expresa en tiroides, pulmón y cerebro anterior; en modelos animales la ausencia de TTF–1 causa agenesia tiroidea y alteraciones cerebrales y pulmonares. En la glándula tiroides adulta este factor controla la síntesis basal de la Tg, TPO, NIS y el receptor a TSH. A la fecha se han descrito ocho mutaciones distintas en humanos para este factor.
2. TTF2 (también conocido como FOXE1, FKHL15 y TITF2). Las mutaciones homocigotas de TTF–2 en animales producen la muerte dentro de las primeras 48 horas de vida. Las mutaciones en humanos se relacionan con agenesia tiroidea, paladar hendido y atresia de las coanas (síndrome de Bamforth–Lazarus). En la glándula adulta este factor interviene en la expresión de Tg y TPO. A la fecha se han descrito dos mutaciones diferentes en humanos.
3. PAX8. Las mutaciones en este factor se relacionan con ectopia o hipoplasia tiroidea. Por el momento se desconoce porqué una mutación heterocigota es suficiente para causar hipotiroidismo en humanos, mientras que en ratones este tipo de mutación no presenta fenotipo anormal. Se ha propuesto la penetrancia incompleta como mecanismo para explicar el heteromorfismo de estos fenotipos; aunque no puede descartarse una modulación por genes modificadores. En el adulto este factor controla la expresión de Tg, NIS y TPO en respuesta a la estimulación por TSH. Tomando en cuenta el tipo de transmisión y la tasa de frecuencia de mutaciones reportadas a la fecha, la probabilidad de que los pacientes con alteraciones en el gen de PAX8 transmitan la enfermedad al 50% de sus hijos es de 0.01. Se han reportado a la fecha ocho diferentes mutaciones de PAX8 en humanos.
Disminución en la acción de las HT (DAHT)
Las acciones de las HT están mediadas por varios receptores nucleares generados por edición (splicing) alternativa de los genes que los codifican: α (RHT–α) y β (RHT–β). La DAHT implica una situación de hipotiroidismo celular intrínseco, algunas veces asociada con eutiroidismo bioquímico o incluso con hipertiroidismo leve. La DAHT siempre es de carácter parcial y se han descrito más de 600 casos, lográndose identificar alteraciones en el gen del RHT–β en cerca del 90%, con 86 mutaciones distintas reportadas hasta la fecha. En la práctica clínica estos neonatos pueden identificarse por la persistente elevación de tironinas circulantes y una concentración de TSH paradójicamente normal o incluso elevada. El espectro clínico de la DAHT puede variar desde anormalidades bioquímicas aisladas hasta una constelación de hallazgos que incluyen bocio, datos de hiper e hipotiroidismo, estatura corta, maduración ósea retardada y alteraciones conductuales. De esta forma, mientras que los pacientes asintomáticos no requieren tratamiento, en un subgrupo de ellos será necesario instaurarlo para aminorar las manifestaciones de hiper o hipotiroidismo.44,71–73
Para que las HT ejerzan su acción se requiere que sean transportadas al interior de la célula y que sean desyodadas. El transporte es mediado por un grupo de proteínas transmembranales identificadas recientemente, siendo las más específicas la OATP1C1 y la MCT8. La OATP1C1 parece ser responsable del transporte de T4 a través de la barrera hematoencefálica, mientras que a la MCT8 se le atribuye el transporte de Tg al interior de las neuronas. A pesar de que no se han identificado alteraciones relacionadas con la OATP1C1, la asociación de niveles circulantes elevados de HT y retraso mental en pacientes con mutaciones en el gen que codifica para la MCT8 sugiere la importancia fisiológica de estos transportadores y su relación con la DAHT.74–77
El diagnóstico diferencial de la DAHT debe hacerse tomando en cuenta aquellas alteraciones en la afinidad de las HT a sus proteínas transportadoras séricas. En este último tipo de alteraciones los niveles totales de HT circulantes generalmente se encuentran elevados, mientras que la TSH está normal; sin embargo, los niveles circulantes de HT libre se mantienen normales. La α–globulina transportadora de tiroxina (TBG), principal proteína transportadora de las HT, es responsable de la mayor parte de estas alteraciones, presentando una forma de transmisión ligada al cromosoma X. Característicamente, los defectos en el transporte sérico de HT no cursan con manifestaciones clínicas; sin embargo, el no reconocer esta alteración oportunamente puede conducir a un tratamiento inapropiado.71,78,79
Dishormonogénesis
1. Deficiencia en la respuesta a TSH. Las alteraciones que conducen a una deficiencia en la respuesta a TSH pueden ser secundarias tanto a mutaciones en el receptor a esta hormona (RTSH), como por alteraciones en su vía de señalización. El RTSH se localiza en la membrana plasmática del tirocito y es una glucoproteína de 744 aminoácidos acoplada a proteínas G con siete dominios transmembranales. Es importante hacer notar que las mutaciones en el RTSH pueden producir fenotipos variables, incluso dentro de una misma familia. Lo anterior sugiere que, tanto el genotipo como influencias ambientales, pueden estar modificando el fenotipo de estos pacientes. A la fecha se han descrito 43 mutaciones homocigotas o heterocigotas compuestas para este receptor.65,80,81 Tomando en cuenta el grado de respuesta funcional, las mutaciones en el RTSH pueden ser clasificadas como parciales o completas. En la resistencia parcial los niveles circulantes de TSH se encuentran elevados, pero con niveles circulantes normales de HT; este cuadro es conocido como hipertirotropinemia eutiroidea. En estos pacientes el tamaño de la glándula tiroides puede ser normal o estar aumentado. Por otra parte, las resistencias completas cursan con hipotiroidismo franco e hipoplasia tiroidea. Debido a la ausencia de captación del trazador en los estudios de gammagrafía, inicialmente algunos de estos pacientes pueden erróneamente diagnosticarse como agenesia tiroidea; sin embargo, una ecosonografía cuidadosa puede revelar la presencia del tejido tiroideo hipoplásico.23,66,71
Los defectos en las proteínas G involucradas en la señal de transducción del RTSH han sido reconocidos como causa de seudohipoparatiroidismo (PHP). Específicamente, el PHPla cursa no sólo con resistencia a la paratohormona, sino con resistencia a la TSH, LH y FSH.82,83
2. Defectos en el transporte del yodo. Durante la síntesis de las HT, el yoduro es transportado activamente en los dos polos funcionales del tirocito: la membrana basolateral y la apical. El transporte en la membrana basolateral facilita el paso del I – de la circulación al interior del tirocito; este paso está mediado por el transportador Na+/r (NIS). El NIS es una proteína de 643 aminoácidos con 13 dominios transmembranales que actúa como simportador, internalizando Na+ y I – hacia el tirocito. Hasta el momento se han descrito 10 mutaciones homocigotas o heterocigotas diferentes para el NIS. Los neonatos con defectos en el NIS presentan hipotiroidismo asociado a disminución en la captación glandular de radio–yodo. Estos pacientes pueden presentar bocio difuso o nodular, aunque en la mayoría de los casos la glándula es de tamaño normal y el bocio aparece más tarde.61,65,71,83 Algunos pacientes con mutaciones inactivantes bialélicas presentan solamente una alteración parcial en el transporte del yoduro, manifestándose clínicamente como bocio eutiroideo. Lo anterior sugiere la presencia de transportadores adicionales no identificados, o bien la entrada del yoduro plasmático hacia la tiroides por difusión.44
Aunque recientemente se ha propuesto la participación de otros transportadores en la membrana apical del tirocito,84 la transferencia del yoduro en este polo funcional del tirocito está mediada al menos parcialmente por el transportador I–/Cl– también conocido como pendrina o PDS. La PDS es una proteína de 780 aminoácidos con 12 dominios transmembranales que actúa como un antiportador, transportando I – fuera del tirocito hacia el coloide a la vez que internaliza Cl –. La entidad clínica relacionada con mutaciones en el gen de la PDS se conoce como síndrome de Pendred. Este síndrome fue inicialmente descrito en 1896 y el cuadro clínico se caracteriza por sordera congénita y bocio; asimismo, estos pacientes pueden presentar una prueba positiva de descarga de yodo ante la administración de perclorato.24,61,85 La prueba del perclorato se utiliza para detectar defectos en la organificación intratiroidea del yodo (vide infra), y su empleo en el diagnóstico del síndrome de Pendred ilustra claramente el papel fisiológico de la PDS. Esta prueba se basa en los siguientes dos principios:
• Ante el cese en el transporte activo de yodo del vaso sanguíneo hacia el interior del tirocito (inhibición del NIS producida con el perclorato), se produce un eflujo de yodo en sentido contrario, siendo esto favorecido por un gradiente de concentración y eléctrico.
• El yodo intratiroideo no organificado (yodo libre) es rápidamente unido a la Tg, de esta forma, el yodo que abandona el tirocito ante la estimulación con perclorato es únicamente el yodo libre. Con base en lo anterior, una disminución significativa en la concentración de radioyodo intratiroideo que sigue a la administración del perclorato, podría indicar la presencia de un defecto en la organificación del yodo.
En una prueba estándar primeramente se administra el radioyodo, cuantificándose dos horas después la radiactividad presente en la glándula tiroides (detección epitiroidea). A continuación se administra el perclorato y se cuantifica nuevamente a las dos horas la radiactividad remanente en la glándula. Una disminución en la radiactividad remanente > 5% sugiere un defecto en el transporte apical del yodo o en su organificación. Frecuentemente los pacientes afectados con el síndrome de Pendred pueden desarrollar un bocio moderado, son usualmente eutiroideos (a menos de que la ingesta de yodo sea baja) y muestran sólo una descarga parcial de yoduro ante la administración de perclorato. El síndrome de Pendred es probablemente la causa más común de sordera asociada a un síndrome, ya que se ha identificado en 10% de los pacientes con sordera hereditaria; sin embargo, la sordera puede no estar presente en todos los casos, y es causada por una alteración en el desarrollo de la cóclea llamada cóclea de Mondini. A la fecha se han descrito 108 mutaciones distintas para el gen de la PDS.61,65,69,86
3. Defectos en la síntesis/degradación de Tg. La Tg es una glucoproteína homodimérica de 2,749 aminoácidos que es producida por el tirocito y secretada hacia el coloide. Además de su función como soporte o matriz para la síntesis de HT, la Tg es el principal almacén de yodo y HT del organismo, lo cual puede ser muy importante durante situaciones de escaso aporte/consumo dietético del halógeno. Una vez sintetizadas, las cadenas nacientes del péptido de Tg inicialmente se agregan, para separarse posteriormente en monómeros que se pliegan y forman dímeros estables. Este proceso es auxiliado por al menos siete proteínas o chaperonas que escoltan a la Tg hacia el Golgi para su posterior glicosilación, sulfatación y fosforilación. Estas modificaciones postraduccionales parecen jugar un papel clave para las señales que le permitirán a la Tg ser transportada a la membrana apical, ser yodada y, posteriormente, endocitada para liberar a las HT de su estructura. La molécula de Tg contiene 66 tirosinas, pero solamente un pequeño número de éstas son yodadas. Esta yodación ocurre en los llamados sitios hormonogénicos localizados en los extremos amino y carboxilo de la proteína. De estos sitios hormonogénicos depende la correcta estructura terciaria del péptido.27,28,61,87
Además de las funciones ya descritas, recientemente se ha propuesto que la Tg regula otros procesos de la hormonogénesis tiroidea. La administración de Tg en concentraciones fisiológicas a células en cultivo, aumenta la proteína y el ARNm de la PDS y, simultáneamente, disminuyen (proteína y ARNm) la propia Tg, la TPO, el NIS y el RTSH.88,89 Igualmente interesante es la propuesta de que la proteólisis de la Tg y la subsiguiente liberación de las HT contenidas en su estructura, inicia en la cavidad folicular, aun antes de la endocitosis, continuando en el interior de los fagolisosomas.90 Se han relacionado diversas cisteinproteasas lisosomales (i.e., catepsinas B, K y L) para esta proteólisis extracelular. La importancia fisiológica de este proceso se constata en la reducción de HT circulantes que exhiben los ratones deficientes de estas cisteinproteasas.91,92
Los defectos en la síntesis/degradación de la Tg implican tanto problemas de cantidad como de calidad en su síntesis. Se ha corroborado que el acoplamiento de MIT y DIT puede estar disminuido de forma importante en individuos con anormalidades estructurales de la Tg. El cuadro clínico es variable y refleja el grado de severidad del defecto; frecuentemente se presenta bocio de dimensiones considerables, pudiéndose encontrar síntomas causados por compresión de estructuras adyacentes en el cuello. Los pacientes pueden cursar con hipotiroidismo subclínico o florido, o incluso ser eutiroideos, presentando en todos los casos una captación de radioyodo elevada. Se han reportado 13 mutaciones diferentes para la Tg en humanos.63,65
4. Alteraciones en la yodación. Las alteraciones en la yodación de la Tg pueden ser resultado tanto de defectos en la TPO como en el sistema generador de H2O2. En este tipo de defectos la producción de HT está disminuida, mientras que la síntesis de Tg y la captación del I" están fuertemente estimuladas por la TSH. El incremento en la síntesis de la Tg puede reflejarse en elevadas concentraciones circulantes de esta proteína. En estos pacientes la captación de radioyodo por la tiroides es alta; sin embargo, el bloqueo en la yodación produce un incremento en la concentración intracelular de yoduro libre. Esto puede verse reflejado en la cantidad de radioyodo liberado por la glándula después de la administración de perclorato (véase sección de defectos en el transporte del yodo). De esta forma los defectos en la yodación pueden ser totales o parciales; los primeros se caracterizan por una descarga de radioyodo > al 90%, mientras que los segundos lo hacen por una descarga > al 10–20% del radioisótopo acumulado. Se considera que los defectos totales en la organificación ocurren aproximadamente en uno de cada 66,000 neonatos, y que la mayoría de éstos presentan un defecto en el gen que codifica para la TPO.60,63,87 La TPO es una hemoproteína glicosilada que está ligada a la membrana apical del tirocito con su sitio catalítico expuesto a la cavidad folicular. Las alteraciones en esta proteína se encuentran entre las causas más frecuentes de HNe debidas a dishormonogénesis, reportándose a la fecha 40 diferentes mutaciones para este gen.60,65 Las reacciones catalizadas por la TPO dependen del H2O2 como cofactor esencial, por lo que, la generación de H2O2 es un punto de control clave en la hormonogénesis. Se han identificado dos oxidasas denominadas THOX1 y THOX2, las cuales presentan siete dominios transmembranales y sitios de unión para NADPH, FAD y Ca2+. Ambas oxidasas se expresan en la membrana apical del tirocito y colocalizan con la TPO. Sin embargo, solamente la THOX2 se ha identificado como factor causal de alteraciones en la yodación en algunos pacientes, reportándose cuatro mutaciones distintas.24,65,71,93
5. Deficiencia en el reciclaje del yodo. Una vez que la Tg madura sufre proteólisis, se liberan de su estructura dos clases de compuestos yodados: las HT y las yodotirosinas que no fueron acopladas (que comprenden 60% del I – contenido en la Tg). Las yodotirosinas son desyodadas por la deshalogenasa tiroidea, enzima también conocida como tDh, lo cual permite que el I – liberado pueda ser reutilizado para un nuevo ciclo de síntesis hormonal. La tDh es una flavoproteína que cataliza la deshalogenación reductiva de MIT y DIT, empleando NADPH como cofactor.94,95 Clínicamente, los pacientes con alteraciones en la tDh pueden presentar hipotiroidismo severo, retraso mental, bocio y concentraciones circulantes elevadas de TSH y yodotirosinas, con la aparición de estas últimas en orina.96,97 Puesto que el defecto se encuentra en el reciclaje intratiroideo del I –, la administración de éste en cantidades suficientes puede compensar su pérdida y establecer así un estado eutiroideo. Recientemente se clonó el gen que codifica para la tDh en humanos, identificándose la proteína en la membrana apical del tirocito.98,99
La bioactividad de las HT depende de su desyodación periférica a nivel de los órganos blanco. La remoción de los átomos de yodo de la molécula de HT es secuencial y está catalizada por selenoenzimas denominadas genéricamente desyodasas de yodotironinas o IDs. Esta desyodación es órgano–específica, y dependiendo del anillo desyodado, se reconoce la vía de activación que produce las formas activas (T3 y 3,5–T2) y la vía de inactivación que produce las formas inactivas (rT3 y 3,3'–T2) de las HT. Con base en sus características bioquímicas y operacionales se distinguen tres isotipos de esta familia enzimática: ID1, ID2 e ID3. La ID1 cataliza tanto la vía de activación como la de inactivación, la ID2 la vía de activación y la ID3 exclusivamente la vía de inactivación.15 En este contexto y en relación con el tema específico de esta revisión, recientemente se ha descrito un cuadro de hipotiroidismo de tipo consuntivo en infantes con hemangioma hepático, donde la sobreexpresión de la ID3 produce depleción de la poza hormonal circulante de HT. En estos pacientes, y a pesar de la hiperestimulación por parte de la TSH endógena, la inactivación hormonal sobrepasa a la producción tiroidea de HT. El manejo de estos pacientes requiere la inmediata administración de grandes dosis de HT y un seguimiento cuidadoso de los niveles circulantes de yodotironinas y TSH. Este tipo de hemangiomas generalmente se presenta dentro del primer año de vida, aunque se ha reportado un caso en adultos.100,102
TRATAMIENTO
Esta sección no pretende revisar detalladamente los lineamientos terapéuticos del HNe, ello rebasa el propósito central de este trabajo. Sin embargo, para el manejo adecuado e integral del neonato con HNe, existen algunas observaciones recientes que merecen ser mencionadas.
Es indudable que independientemente de su etiología y tan pronto como el diagnóstico de HNe sea establecido, se debe iniciar el tratamiento sustitutivo con HT. En este sentido, tradicionalmente el HNe se ha manejado mediante la administración combinada de T4 y T3. Sin embargo, ahora se reconoce que la suplementación con T4 es suficiente puesto que los niveles circulantes de T3 se normalizan a los 2 o 3 días de iniciado el tratamiento.103,104
Es importante señalar que se pueden presentar ciertas dificultades en el aprendizaje y desarrollo psicomotor incluso en el neonato tratado precozmente. En un estudio reciente se analizaron 106 niños afectados con HNe (diagnosticados por tamizaje neonatal), encontrándose una disminución en el coeficiente de inteligencia de entre 6 a 8 puntos, al ser comparados con infantes consanguíneos de edad similar. Lo anterior sugiere fuertemente que los niveles adecuados de HT durante el desarrollo embrionario son críticos para un desarrollo neurológico normal.23,34,105,108 Tradicionalmente, el objetivo de la terapia sustitutiva ha sido incrementar la concentración circulante de T4 y normalizar las concentraciones circulantes de TSH. Sin embargo, existen reportes que indican que el desarrollo del neonato con HNe no es homogéneo. Aún más, se ha encontrado que el coeficiente intelectual en la evolución de un niño con HNe depende de variables como la severidad de la deficiencia, el tiempo de inicio del tratamiento, la dosis de T4 administrada y los niveles circulantes de HT a lo largo del tratamiento. Por esta razón, actualmente se considera que los niños con agenesia de la glándula tiroides necesitan una evaluación más cuidadosa y un esquema de tratamiento más intenso.109,110 Por otra parte, en niños con dishormonogénesis, la estimulación subsiguiente por parte de la TSH puede ser un factor de riesgo para el desarrollo posterior de hiperplasia nodular. Consecuentemente, en estos pacientes se recomienda un seguimiento más cercano a edades mayores.111,112
La Academia Americana de Pediatría recomienda dosis iniciales de tiroxina de 10 a 15 µg/kg/día para el infante con T4 baja y TSH elevada. Sin embargo, la tendencia actual recomienda iniciar con una dosis mayor de tiroxina.103,113,115 En este sentido, es importante tener en mente que el neonato normal presenta niveles más altos de T4 circulante en los primeros meses de vida, a comparación del adulto. Por otra parte, es igualmente importante prevenir el sobretratamiento del neonato afectado, ya que los efectos adversos incluyen craneosinostosis, aceleración del crecimiento y maduración esquelética y problemas de temperamento y conducta.37
Por otra parte, se ha documentado una marcada correlación positiva entre la alta morbimortalidad observada en el recién nacido prematuro y la hipotiroxinemia. Por esta razón, se ha propuesto el uso de T4 para mejorar el pronóstico del prematuro con hipotiroxinemia que cursa con niveles circulantes normales de TSH y que no presenta alteraciones hipotálamo–hipofisiarias. Sin embargo, se desconoce si realmente la hipotiroxinemia es causa o efecto de las alteraciones observadas en el neonato prematuro. Aún más, la opinión actual considera que los estudios existentes no presentan evidencia concluyente acerca de un posible efecto benéfico del tratamiento con HT en los infantes prematuros con hipotiroxinemia, razón por la cual esta práctica no se recomienda.116–118
PERSPECTIVAS
La importancia de la detección temprana del HNe y del tamizaje neonatal como auxiliar diagnóstico es innegable. Basadas en este conocimiento, las políticas de salud pública y vigilancia epidemiológica han permitido la detección y tratamiento oportunos del HNe en los países del primer mundo. En México no debe aplazarse más la concertación de esfuerzos interinstitucionales para que toda la población quede incluida en este tipo de políticas. Por otra parte, y dado el potencial disruptivo que los organohalógenos industriales tienen en la función tiroidea, es necesario iniciar el estudio sistemático de estos agentes en nuestro país.
En la actualidad no basta con realizar el diagnóstico e instaurar el tratamiento del HNe; es necesario también identificar su etiología con la finalidad de individualizar el tratamiento y pronóstico del neonato afectado. Sin embargo, en un gran número de pacientes el defecto aún no puede ser identificado a nivel molecular. En efecto, estudios recientes muestran que la mayoría (70%) de los ARNm expresados en el tirocito normal, todavía no pueden ser identificados.119 Estos hallazgos exhiben la ignorancia que aún prevalece en la comprensión de la fisiología tiroidea. Igualmente, ellos muestran la necesidad de profundizar en el estudio bioquímico y molecular de los sistemas tiroideos. Es este conocimiento el que permitirá comprender la fisiopatología del HNe, y desarrollar así nuevas estrategias para prevenir o tratar esta enfermedad.
AGRADECIMIENTOS
Agradecemos a todos aquellos investigadores listados en las referencias, cuyas contribuciones fueron invaluables en la preparación de esta revisión. Igualmente agradecemos al Dr. Jorge Larriva S, al Dr. Manuel Salas A, M en C Patricia Villalobos A, y a la Dra. Marcela Vela por sus críticos y enriquecedores comentarios a este trabajo. Agradecemos también la invaluable asistencia hemerográfica de Pilar Galarza (INB–UNAM).
REFERENCIAS
1. Gómez VE, Bolaños F, Valverde RC. Tiroides. En: Malacara JM, García VM, Valverde RC (Eds.). Fundamentos de endocrinología. México: Salvat; 1990. [ Links ]
2. Brent GA. Tissue–specific sctions of thyroid hormone: insights from animal models. Rev Endocr Metab Disord 2000; 1(1): 27–33. [ Links ]
3. Anderson GW, Mariash CN, Oppenheimer JH. Molecular actions of thyroid hormone. En: Braverman LE, Utiger RD (eds.). The thyroid: a fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, p. 977–83. [ Links ]
4. Feng X, Jiang Y, Meltzer P, et al. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol Endocrinol 2000; 14(7): 947–55. [ Links ]
5. Yen PM. Cellular action of thyroid hormone. Chapter 3. The thyroid and its diseases– Book chapters. 2003 (fecha de acceso: febrero 2006). Disponible en: http://www.thyroidmanager.org/Chapter3/3d–frame.htm [ Links ]
6. Bernal J, Guadaño–Ferraz A, Morte B. Perspectives in the study of thyroid hormone action on brain development and function. Thyroid 2003; 13(11): 1005–12. [ Links ]
7. Chidakel A, Mentuccia D, Celi FS. Peripheral metabolism of thyroid hormone and glucose homeostasis. Thyroid 2005; 15(8): 899–903. [ Links ]
8. Pop VJ, Kuijpens JL, van Baar AL, et al. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol 1999; 50(2): 149–55. [ Links ]
9. New England congenital hypothyroidism collaborative. Correlation of cognitive test scores and adequacy of treatment in adolescents with congenital hypothyroidism. J Pediatr 1994; 124(3): 383–7. [ Links ]
10. Frieden E. The chemical elements of life. Set Am 1972; 227: 52–60. [ Links ]
11. Mertz W. The essential trace elements. Science 1981; 213: 1332–8. [ Links ]
12. Bloomfield MM, Stephens LJ. Chemistry and the living organisms. New York: John Wiley & Sons; 1996. [ Links ]
13. Gribble GW. Naturally occurring organohalogen compounds. Accts Chem Res 1998; 31: 141–52. [ Links ]
14. Emsley J. Nature's building blocks. New York: Oxford University Press; 2001. [ Links ]
15. Valverde–R C, Orozco A, Becerra A, et al. Halometabolites and cellular dehalogenase systems: an evolutionary perspective, Int Rev Cytol 2004; 234: 143–99. [ Links ]
16. Gribble GW. The diversity of naturally produced organohalogens. Chemosphere 2003; 52: 289–97. [ Links ]
17. Burman KD, Wartofsky L. Iodine effects on the thyroid gland: biochemical and clinical aspects. Rev Endocr Metab Disord 2000; 1(1): 19–25. [ Links ]
18. Larsen PR, Davies TF, Schlumberger MJ, Hay ID. Thyroid physiology and diagnostic evaluation of patients with thyroid disorder. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS (eds.). Williams textbook of endocrinology. Philadelphia: Saunders; 2003, p. 331–73. [ Links ]
19. Pintar JE. Normal development of the hypothalamic–pituitary–thyroid axis. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, p. 7–19. [ Links ]
20. Davies TF, Ando T, Lin RY, et al. Thyrotropin receptor–associated diseases: From adenomata to Graves disease. J Clin Invest 2005; 115(8): 1972–83. [ Links ]
21. Missero C, Cobellis G, De Felice M, et al. Molecular events involved in differentiation of thyroid follicular cells. Mol Cell Endocrinol 1998; 140(1–2): 37–43. [ Links ]
22. Moore KL. Aparato branquial, cabeza y cuello. En: Moore KL (ed.). Embriología clínica. México: Interamericana; 1989, pp. 187–227. [ Links ]
23. Radetti G, Zavallone A, Gentili L, et al. Foetal and neonatal thyroid disorders. Minerva Pediatr 2002; 54(5): 383–400. [ Links ]
24. Dunn JT, Dunn AD. Update on intrathyroidal iodine metabolism. Thyroid 2001; 11(5): 407–14. [ Links ]
25. Kim PS, Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: Disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev 1998; 19(2): 173–202. [ Links ]
26. Carrasco N. Thyroid iodide transport: the Na+/I– symporter (NIS). In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 52–61. [ Links ]
27. Taurog AM. Hormone synthesis: Thyroid iodine metabolism. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 61–85. [ Links ]
28. Dunn JT, Dunn AD. Thyroglobulin: Chemistry, biosynthesis and proteolysis. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 91–104. [ Links ]
29. Andersson HC, Kohn LD, Bernardini I, et al. Characterization of lysosomal monoiodotyrosine transport in rat thyroid cells. Evidence for transport by system H. J Biol Chem 1990; 265(19): 10950–4. [ Links ]
30. Snyder PJ. The pituitary in hypothyroidism. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 811–14. [ Links ]
31. Silva EJ. Catecholamines and the sympathoadrenal system in hypothyroidism. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 820–3. [ Links ]
32. Diekman T, Lansberg PJ, Kastelein JJ, et al. Prevalence and correction of hypothyroidism in a large cohort of patients referred for dyslipidemia. Arch Inter Med 1995; 155(14): 1490–5. [ Links ]
33. Franklyn JA. Metabolic changes in hypothyroidism. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 833–6. [ Links ]
34. Foley TP. Congenital hypothyroidism. Hypothyroidism in infants and children. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 977–83. [ Links ]
35. Grant DB, Smith I, Fuggle PW, et al. Congenital hypothyroidism detected by neonatal screening: Relationship between biochemical severity and early clinical features. Arch Dis Child 1992; 67(1): 87–90. [ Links ]
36. Unachak K, Dejkhamron P. Primary congenital hypothyroidism: Clinical characteristics and etiological study. J Med Assoc Thai 2004; 87(6): 612–7. [ Links ]
37. LaFranchi S. Congenital hypothyroidism: Etiologies, diagnosis, and management. Thyroid 1999; 9(7): 735–40. [ Links ]
38. Brown R, Larsen PR. Thyroid gland development and disease in infancy and childhood. Chapter 15. The thyroid and its diseases – Book chapters. 2005 (fecha de acceso: febrero 2006). Disponible en: http://www.thyroidmanager.org/Chapter15/15-frame.htm [ Links ]
39. Rovet JF, Ehrlich RM. Long–term effects of L–thyroxine therapy for congenital hypothyroidism. J Pediatr 1995; 126(3): 380–6. [ Links ]
40. Glorieux J, Dussault J, Van Vliet G. Intellectual development at age 12 years of children with congenital hypothyroidism diagnosed by neonatal screening. J Pediatr 1992; 121(4): 581–4. [ Links ]
41. Dussault JH, Coulombe P, Laberge C, et al. Preliminary report on a mass screening program for neonatal hypothyroidism. J Pediatr 1975; 86: 670–4. [ Links ]
42. Poder–Ejecutivo–Federal. Norma técnica No 321 para la prevención del retraso mental producido por hipotiroidismo congénito. México: Gobierno Constitucional de los Estados Unidos Mexicanos. México: Diario Oficial de la Federación 1988; 14: 88–90. [ Links ]
43. Vela–Amieva M, Hernández–Osorio C, Gamboa–Cardiel S, et al. Hipertirotropinemia en recién nacidos Mexicanos. Salud Pública Mex 2003; 45(4): 269–75. [ Links ]
44. Moreno JC, De Vijlder JJ, Vulsma T, et al. Genetic basis of hypothyroidism: recent advances, gaps and strategies for future research. Trends Endocrinol Metab 2003; 14(7): 318–26. [ Links ]
45. Hetzel BS. Iodine Deficiency Disorders (IDD), and their eradication, lancet 1983; 2: 1126–9. [ Links ]
46. World Health Organization. Assessment of iodine deficiency disorders and monitoring their elimination. A guide for programme managers. Second edition, 2001; WHO/NHD/01.1. [ Links ]
47. Delange FM, Hetzel B. The iodine deficiency disorders. Chapter 20. The thyroid and its diseases – Book chapters. 2005 (fecha de acceso: febrero 2006). Disponible en: http://www.thyroidmanager.org/Chapter20/20-frame.htm [ Links ]
48. Kavishe FP. Iodine deficiency disorders. In: Sadler MJ, Strain JJ, Caballero B (eds.). Encyclopedia of Human Nutrition. Vol. 3. London: Academic Press; 1999, pp. 1146–53. [ Links ]
49. Vitti P, Rago T, Aghini–Lombardi F, et al. Iodine deficiency disorders in Europe. Public Health Nutrition 2001; 4(2B): 529–35. [ Links ]
50. World Health Organization. Iodine deficiency disorders. WHO fact sheet No 121. Geneva, Switzerland: WHO; 1996. [ Links ]
51. Hetzel BS. Eliminating iodine deficiency disorders: The role of the international council in the global partnership. Bull World Health Organ 2002; 80(5): 410–12. [ Links ]
52. WHO, UNICEF, ICCIDD. Progress towards elimination of iodine deficiency disorders. Geneva: World Health Organization; 1999. [ Links ]
53. Vela M, Gamboa S, Loera–Luna A, et al. Neonatal screening for congenital hypothyroidism in México: experience, obstacles, and strategies. J Med Screen 1999; 6: 77–9. [ Links ]
54. Vela–Amieva M, Gamboa–Cardiel S, Pérez–Andrade ME, et al. Epidemiología del hipotiroidismo congénito en México. Salud Pública Mex 2004; 46(2): 141–8. [ Links ]
55. Delange FM. Endemic cretinism. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 743–54. [ Links ]
56. Martínez–Salgado H, Castañeda–Limones R, Lechuga–Martín del Campo D, et al. Deficiencia de yodo y otros posibles bociógenos en la persistencia del bocio endémico en México. Gac Méd Méx 2002; 138(2): 149–56. [ Links ]
57. Vásquez–Garibay EM, Romero–Velarde E, Nápoles–Rodriguez F, et al. Prevalencia de deficiencia de hierro y yodo, y parasitosis en niños de Arandas, Jalisco, México. Salud Pública Mex 2002; 44(3): 195–200. [ Links ]
58. Klein RZ, Mitchell ML. Hypothyroidism in infants and children. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 973–7. [ Links ]
59. Fugazzola L, Persani L, Mannavola D, et al. Recombinant human TSH testing is a valuable tool for differential diagnosis of congenital hypothyroidism during L–thyroxine replacement. Clin Endocrinol 2003; 59(2): 230–6. [ Links ]
60. De Vijlder JJ, Vulsma T. Hereditary metabolic disorders causing hypothyroidism. In: Braverman LE, Utiger RD (Eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 733–42. [ Links ]
61. Kopp P. Pendred's syndrome and genetic defects in thyroid hormone synthesis. Rev Endocr Metab Disord 2000; 1: 109–21. [ Links ]
62. McKusick VA. Mendelian inheritance in man. A catalog of human genes and genetic disorders. Baltimore: Johns Hopkins University Press; 1998. [ Links ]
63. Vono–Toniolo J, Kopp P. Thyroglobulin gene mutations and other genetic defects associated with congenital hypothyroidism. Arq Bras Endocrinol Metab 2004; 48(1): 70–82. [ Links ]
64. Vassart G. TSH receptor mutations and diseases. Chapter 16. The thyroid and its diseases – Book chapters. 2003 (fecha de acceso: febrero 2006). Disponible en: http://www.thyroidmanager.org/Chapter16/16a-frame.htm [ Links ]
65. Stenson PD, Ball EV, Mort M, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat 2003; 21(6): 577–81 (véase también: http://www.hgmd.cf.ac.uk/). [ Links ]
66. Macchia PE, De Felice M, Di Lauro R. Molecular genetics of congenital hypothyroidism. Curr Opin Genet Dev 1999; 9(3): 289–94. [ Links ]
67. De Felice M, Ovitt C, Biffali E, et al. A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat Genet 1998; 19(4): 395–8. [ Links ]
68. Tell G, Pellizzari L, Esposito G, et al. Structural defects of a Pax8 mutant that give rise to congenital hypothyroidism. Biochem J 1999; 341(Pt 1): 89–93. [ Links ]
69. Macchia PE. Recent advances in understanding the molecular basis of primary congenital hypothyroidism. Mol Med Today 2000; 6(1): 36–42. [ Links ]
70. Gillam MP, Kopp P. Genetic regulation of thyroid development. Curr Opin Pediatr 2001; 13(4): 358–63. [ Links ]
71. Kopp P. Perspective: Genetic defects in the etiology of congenital hypothyroidism. Endocrinology 2002; 143(6): 2019–24. [ Links ]
72. Refetoff S. Resistance to thyroid hormone. In: Braverman LE, Utiger RD (eds.). The thyroid: A fundamental and clinical text. New York: Lippincott Williams & Wilkins; 2000, pp. 1028–43. [ Links ]
73. Refetoff S. Resistance to thyroid hormone. Chapter 16d. The thyroid and its diseases – Book chapters. 2004 (fecha de acceso: febrero 2006). Disponible en: http://www.thyroidmanager.org/Chapter16/16d-frame.htm [ Links ]
74. Friesema E, Grueters A, Halestrap A. et al. Mutations in a thyroid hormone transporter in patients with severe psicomotor retardation and high serum T3 levels. Thyroid 2003; 13(7): 672. [ Links ]
75. Dumitrescu AM, Liao XH, Best TB, et al. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 2004; 74(1): 168–75. [ Links ]
76. Friesema ECH, Jansen J, Visser TJ. Thyroid hormone transporters. Biochem Soc Trans 2005; 33(1): 228–32. [ Links ]
77. Jansen J, Friesema ECH, Milici C, Visser TJ. Thyroid hormone transporters in health and disease. Thyroid 2005; 15(8): 757–68. [ Links ]
78. Weiss RE, Refetoff S. Resistance to thyroid hormone. Reviews in Endocrine and Metabolic Disorders 2000; 1(1): 97–108. [ Links ]
79. Refetoff S. Defects of thyroid hormone transport in serum. Chapter 16c. The thyroid and its diseases – Book chapters. 2004 (fecha de acceso: febrero 2006). Disponible en: http://www.thyroidmanager.org/Chapter16/16c–frame.htm [ Links ]
80. Yen PM. Thyrotropin receptor mutations in thyroid diseases. Rev Endocr Metab Disorde 2000; 1(1): 123–9. [ Links ]
81. Davies TF, Ando T, Lin RY, Tomer Y, et al. Thyrotropin receptor–associated diseases: From adenomata to graves disease. J Clin Invest 2005; 115(8): 1972–83. [ Links ]
82. Spiegel AM. Mutations in G proteins and G protein–coupled receptors in endocrine disease. J Clin Endocrinol Metab 1996; 81(7): 2434–42. [ Links ]
83. Gillam MP, Kopp P. Genetic defects in thyroid hormone synthesis. Curr Opin Pediatr 2001; 13(4): 364–72. [ Links ]
84. Rodriguez AM, Perron B, Lacroix L, et al. Identification and characterization of a putative human iodide transporter located at the apical membrane of thyrocytes. J Clin Endocrinol Metab 2002; 87(7): 3500–3. [ Links ]
85. Yoshida A, Taniguchi S, Hisatome I, et al. Pendrin is an iodide–specific apical porter responsible for iodide efflux from thyroid cells. J Clin Endocrinol Metab 2002; 87(7): 3356–61. [ Links ]
86. Franklyn J, Shephard M. Evaluation of thyroid function in health and disease. Chapter 6e. The thyroid and its diseases –Book chapters. 2000 (fecha de acceso: febrero 2006). Disponible en: http://www.thyroidmanager.org/Chapter6/6–text.htm [ Links ]
87. De Vijlder JJ. Primary congenital hypothyroidism: Defects in iodine pathways. Eur J Endocrinol 2003; 149(4): 247–56. [ Links ]
88. Suzuki K, Lavaroni S, Mori A, et al. Autoregulation of thyroid–specific gene transcription by thyroglobulin. Proc Nati Acad Sci 1998; 95(14): 8251–6. [ Links ]
89. Suzuki K, Nakazato M, Ulianich L, et al. Thyroglobulin autoregulation of thyroid–specific gene expression and follicular function. Rev Endocr Metab Disord 2000; 1(3): 217–24. [ Links ]
90. Brix K, Lemansky P, Herzog V. Evidence for extracellularly acting cathepsins mediating thyroid hormone liberation in thyroid epithelial cells. Endocrinology 1996; 137(5): 1963–74. [ Links ]
91. Linke M, Jordans S, Mach L, et al. Thyroid stimulating hormone upregulates secretion of cathepsin B from thyroid epithelial cells. Biol Chem 2002; 383(5): 773–84. [ Links ]
92. Friedrichs B, Tepel C, Reinheckel T, et al. Thyroid functions of mouse cathepsins B, K, and L. J Clin Invest 2003; 111(11): 1733–45. [ Links ]
93. De Deken X, Wang D, Many MC, et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem 2000; 275(30): 23227–33. [ Links ]
94. Rosenberg IN, Goswami A. Iodotyrosine deiodinase from bovine thyroid. Methods in Enzymology 1984; 107: 488–500. [ Links ]
95. Solis–S JC, Villalobos P, Orozco A, Valverde–R C. Comparative kinetic characterization of rat thyroid iodotyrosine dehalogenase and iodothyronine deiodinase type 1. J Endocrinol 2004; 181(3): 385–92. [ Links ]
96. Stanbury JB, Meijer JWA, Kassenaar AAH. The metabolism of iodotyrosines. II. The metabolism of mono– and diiodotyrosine in certain patients with familial goiter. J Clin Endocrinol Metab 1956; 16: 848–61. [ Links ]
97. Querido A, Stanbury JB, Kassenaar AAH, et al. The metabolism of iodotyrosines. III. Di–iodotyrosine deshalogenating activity of human thyroid tissue. J Clin Endocrinol Metab 1956; 16: 1096–112. [ Links ]
98. Moreno JC, Keijser R, Aarraas S, et al. Cloning and characterization of a novel thyroidal gene encoding proteins with a conserved nitroreductase domain. J Endocrinol Invest 2002; 25: 23. [ Links ]
99. Gnidehou S, Caillou I, Talbot M, et al. Iodotyrosine dehalogenase 1 (DEHAL1) is a transmembrane protein involved in the recycling of iodide close to the thyroglobulin iodination site. FASEB 2004; 18: 1574–6. [ Links ]
100. Huang SA, Tu HM, Harney JW, et al. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med 2000; 343(3): 185–9. [ Links ]
101. Konrad D, Ellis G, Perlman K. Spontaneous regression of severe acquired infantile hypothyroidism associated with multiple liver hemangiomas. Pediatrics 2003; 112(6): 1424–6. [ Links ]
102. Huang SA, Fish SA, Dorfman DM, et al. A 21–year–old woman with consumptive hypothyroidism due to a vascular tumor expressing type 3 iodothyronine deiodinase. J Clin Endocrinol Metab 2002; 87(10): 4457–61. [ Links ]
103. Selva KA, Mandel SH, Rien L, et al. Initial treatment dose of L–thyroxine in congenital hypothyroidism. J Pediatr 2002; 141(6): 786–92. [ Links ]
104. Cassio A, Cacciari E, Cicognani A, et al. Treatment for congenital hypothyroidism: Thyroxine alone or thyroxine plus triiodothyronine? Pediatrics 2003; 111(5 Pt 1): 1055–60. [ Links ]
105. Rovet JF, Ehrlich RM, Sorbara DL. Neurodevelopment in infants and preschool children with congenital hypothyroidism: Etiological and treatment factors affecting outcome. J Pediatr Psychol 1992; 17(2): 187–213. [ Links ]
106. Gottschalk B, Richman RA, Lewandowski L. Subtle speech and motor deficits of children with congenital hypothyroid treated early. Dev Med Child Neurol 1994; 36(3): 216–20. [ Links ]
107. Rovet JF. Congenital hypothyroidism: long–term outcome. Thyroid 1999; 9(7): 741–8. [ Links ]
108. Rovet JF.Children with congenital hypothyroidism and their siblings: do they really differ? Pediatrics 2005; 115(1): 52–6. [ Links ]
109. Hanukoglu A, Perlman K, Shamis I, et al. Relationship of etiology to treatment in congenital hypothyroidism. J Clin Endocrinol Metab 2001; 86(1): 186–91. [ Links ]
110. Oerbeck B, Sundet K, Kase BF, et al. Congenital hypothyroidism: Influence of disease severity and L–thyroxine treatment on intellectual, motor, and school–associated outcomes in young adults. Pediatrics 2003; 112(4): 923–30. [ Links ]
111. Alabbasy AJ, Delbridge L, Eckstein R, et al. Microfollicular thyroid adenoma and congenital goitrous hypothyroidism. Arch Dis Child 1992; 67(10): 1294–5. [ Links ]
112. Aronson R, Sochett E, Pearl RH, et al. Nodular hyperplasia in treated congenital goitrous hypothyroidism. J Pediatr Endocrinol Metab 1996; 9(6): 613–6. [ Links ]
113. Hindmarsh PC. Optimization of thyroxine dose in congenital hypothyroidism. Arch Dis Child 2002; 86(2): 73–5. [ Links ]
114. Hopwood NJ. Treatment of the infant with congenital hypothyroidism. J Pediatr 2002; 141(6): 752–4. [ Links ]
115. Heyerdahl S, Oerbeck B. Congenital hypothyroidism: Developmental outcome in relation to levothyroxine treatment variables. Thyroid 2003; 13(11): 1029–38. [ Links ]
116. Osborn DA. Thyroid hormones for preventing neurodevelopmental impairment in preterm infants. Cochrane Database Syst Rev 2001; (4): CD001070. [ Links ]
117. Ogilvy–Stuart AL. Neonatal thyroid disorders. Arch Dis Child Fetal Neonatal Ed 2002; 87(3): F165–F171. [ Links ]
118. Rapaport R, Rose SR, Freemark M. Hypothyroxinemia in the preterm infant: the benefits and risks of thyroxine treatment. J Pediatr 2001; 139(2): 182–8. [ Links ]
119. Pauws E, Moreno JC, Tijssen M, et al. Serial analysis of gene expression as a tool to assess the human thyroid expression profile and to identify novel thyroidal genes. J Clin Endocrinol Metab 2000; 85: 1923–7. [ Links ]
Abreviaturas: HNe, hipotiroidismo neonatal; HT, hormonas tiroideas; I', ion yoduro; MIT, monoyodotirosina; DIT, diyodotirosina; T4, tetrayodotironina; T3, triyodotironina; TSH, tirotropina u hormona estimulante de la tiroides; NIS, simportador Na/1; PDS, pendrina; Tg, tiroglobulina; BiP, proteína de unión también conocida como proteína 78 regulada por glucosa; TPO, peroxidasa tiroidea; THOX, oxidasa tiroidea; tDh, deshalogenasa tiroidea; GRP94, proteína 94 regulada por glucosa; PDI, proteína disulfuro isomerasa; NADPH, nicotin–adenin dinucleótido de fosfato reducido; IDD, trastornos secundarios a deficiencia de yodo; HiT, hipotiroidismo transitorio; DHH, defectos hipotálamo–hipofisiarios; HESX1, gen tipo homeobox expresado en células ES; LHX3, proteína 3 de humano con homeodominio LIM; PROP1, factor de transcripción tipo homeodominio específico de hipófisis; P0U1F1, factor de transcripción 1 específico de hipófisis; PITX1, panactivador de hormonas hipofisiarias; PITX2, homeobox 2 hipofisiario; GATA2, proteína 2 que se une a secuencias GATA; TRH, hormona liberadora de tirotropina; DisT, disgenesias tiroideas; TTF1, factor de transcripción tiroideo 1; TTF2, factor de transcripción tiroideo 2; PAX8, factor de transcripción 8 de tipo caja pareada; DAHT, disminución en la acción de las HT; RHT, receptor a HT; MCT8, transportador de monocarboxilato 8; OATP1C1, transportador aniónico de tipo orgánico 1C1; RTSH, receptor a TSH; PHP, seudohipoparatiroidismo; LH, hormona luteinizante; FSH, hormona estimuladora del folículo; ARNm, ácido ribonucleico mensajero; FAD, flavin adenin–dinucleótido.

