










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta médica de México
versión On-line ISSN 2696-1288versión impresa ISSN 0016-3813
Gac. Méd. Méx vol.141 no.6 Ciudad de México nov./dic. 2005
Artículo original
El papel de la leptina en el desarrollo de esteatosis hepática y litiasis biliar
Leptine participation in the development of liver steatosis and biliar lithiasis
Nahum Méndez–Sánchez,ª* Guadalupe Ponciano–Rodríguez,ª Norberto C. Chávez–Tapia,ª y Misael Uribeª
ª Departamentos de Investigación Biomédica, Gastroenterología, y Unidad de Hígado. Fundación Clínica Médica Sur, México, D.F., México
*Correspondencia y solicitud de sobretiros:
Dr. Nahum Méndez–Sánchez.
Departamentos de Investigación Biomédica, Gastroenterología, y Unidad de Hígado, Fundación Clínica Médica Sur.
Puente de Piedra 150, Col. Toriello Guerra,
14050 Tlalpan, México, D.F. México.
Correo electrónico: nmendez@medicasur.org.mx
Recibido en su versión modificada: 26 de mayo de 2005
Aceptado: 27 de mayo de 2005
Resumen
La obesidad incrementa el riesgo para varios padecimientos gastro intestinales como litiasis biliar (LB), esteatosis hepática (EH) y este–atohepatitis no alcohólica.
Recientemente observamos una relación entre colesterol HDL, índice de saturación de colesterol en la bilis y leptina en pacientes obesos en reducción de peso. De igual manera la leptina tiene un papel importante en el desarrollo de la EH y probablemente en los mecanismos inflamatorios. El objetivo de este trabajo fue investigar la relación entre la LB y la EH. Se estudiaron a sujetos de la Unidad de Diagnóstico Clínico que acudieron a la realización de una revisión clínica preventiva. Aquellos que presentaron LB o EH por ultrasonido fueron considerados como casos, se compararon con controles sanos. Se tomaron medidas antropométricas, índice de masa corporal (IMC) y concentraciones de leptina, insulina, lípidos séricos, y lipoproteínas por métodos convencionales. Se estudiaron 317 sujetos, quienes fueron divididos en cuatro grupos: LB (n = 100), EH (n = 84), LB + EH (n = 33) y control (n = 100). La edad del grupo control fue significativamente mayor (LB, 52.6 ± 11.6; EH, 49.8 ± 11.1; LB +EH, 51.6 ± 10,5; controles 57.1 ± 7.4), p< 0.05. ElIMC fue mayor en los grupos de EH (28.7 ± 2.8) y LB +EH (29.0 ± 3.8) que en los grupos de LB (27.4 ± 4.3) y control (27.0 ± 3.0), p< 0.05. El grupo de LB (13.7 ± 8.1) presentó las concentraciones más elevadas de leptina comparado con los otros grupos, P < 0.05. Mientras que las concentraciones de insulina fueron similares en los cuatro grupos de sujetos. Los resultados del presente estudio muestran que los sujetos con LB y EH presentan concentraciones elevadas de leptina, comparados con controles. Esto sugiere que la leptina juega un papel importante en la fisiopatología de la LB y EH.
Palabras clave: Litiasis biliar, leptina, insulina, obesidad.
Summary
Obesity increases significantly the risk of developing several common gastrointestinal diseases such as gallstone disease (GD) and hepatic steatosis (HS). Elsewhere we have shown a relationship between HDL cholesterol, cholesterol saturation index, and leptin in obese patients loosing weight. Furthermore, leptin plays an important role facilitating HS and possibly in the associated inflammatory process. The aim of this study was to investigate the relationship between GD and HS. The sample was comprised by patients attending the unit for check up. Subjects with visible stones or HS by ultrasound (cases) were compared with healthy controls. Demographic and body mass index (BMI) were recorded. Plasma leptin, insulin and serum lipids and lipoproteins levels were measured by standard methods. A total of 317 subjects were included in this study. They were divided in four groups as follows: GD (n=100), HS (n=84), GD + HS (n=33) and controls (n=100). The control group was significantly older (GD, 52.6 ± 11.6; HS, 49.8 ±11.1; GD +HS, 51.6 ±10.5; 57.1 ± 7.4), p< 0.05. BMI was higher in the HS groups (28.7 ± 2.8) and GD +EH (29.0 ± 3.8) than in the GD (27.4 ± 4.3) and control (27.0 ± 3.1) group, p< 0.05. The GD group displayed the highest leptin levels (13.7 241 8.1), P < 0.05, whereas insulin levels were similar in all groups. Since GD and HS subjects have high plasma leptin levels compared with controls, our results suggest that leptin plays an important role in the pathophysiology of GD and HS.
Key words: Gallstone, leptin, insulin, obesity.
Introducción
Las encuestas nacionales en EE.UU. y en México1–5 han demostrado incremento paulatino de la obesidad en las últimas décadas.6,7 Las enfermedades relacionadas con la obesidad como la diabetes mellitus, hipertensión arterial, cardiopatía isquémica y síndrome metabólico, entre otras, han sido descritas y analizadas en forma detallada, así como la relación de la obesidad y su impacto con las enfermedades hepatobiliares ha sido descrito recientemente.8,9 El vínculo entre litiasis biliar (LB) y obesidad es conocido desde hace años, debido a que se observa una correlación lineal entre el peso corporal y la secreción de colesterol a nivel biliar.10 Inicialmente Bennion et al,10 y posteriormente StahIberg et al11 demostraron que el principal defecto es un exceso en la síntesis de colesterol y que ni la reducción en la síntesis de sales biliares o la reducida esterificación de colesterol contribuyen al aumento en la secreción de colesterol.
Por otro lado, la obesidad también se encuentra implicada en la patogénesis de la esteatosis hepática (EH), a la cual se debe un porcentaje importante de los casos de cirrosis criptogénica.12 En 1980 Ludwig et al,13 dan el nombre de esteatohepatitis no alcohólica (EHNA) a un síndrome clinicopatológico bien reconocido que se presenta predominantemente en personas con obesidad, del género femenino con diabetes mellitus, en los que no existe el antecedente de consumo y/o abuso de alcohol, pero en la biopsia hepática se observan cambios histopatológicos similares a los que se presentan en hepatitis alcohólica.13
Dependiendo del criterio diagnóstico utilizado la prevalen–cia de EHNA va de 2.8% hasta 25% en la población general,14–16 siendo mucho mayor en grupos de alto riesgo, con valores de entre 70% y 86%, en pacientes obesos y/o diabéticos.17,18
El descubrimiento de la leptina y su papel en la fisiopatología de la obesidad,19 a través del control de la saciedad y del metabolismo energético, ha estimulado el desarrollo de diversos estudios clínicos,20 que intentan establecer el papel de esta hormona en las entidades que acompañan a la obesidad.21 Recientemente informamos que existe una correlación entre colesterol asociado a lipoproteínas de alta densidad (C–HDL), el índice de saturación de colesterol y las concentraciones plasmáticas de leptina en pacientes obesos que se encuentran en reducción de peso.22 Por lo anterior, considerando el papel que tiene la leptina en el desarrollo de LB23 y en la EHNA,24–26 el objetivo de este trabajo fue investigar si existe una relación entre la LB y la EH en donde la leptina juega un papel importante.
Métodos
Población y muestra
Este estudio se llevó a cabo en la Unidad de Diagnóstico Clínico (UDC) de la Fundación Clínica Médica Sur. El estudio se aprobó por el Comité de Ética de la Fundación Clínica Médica Sur conforme la declaración de Helsinki de 1975, y se obtuvo consentimiento informado de todos los participantes. El universo de trabajo se integró con una serie de sujetos asintomáticos que acudieron a la UDC, como parte de una revisión de rutina anual, no por enfermedad sintomática. Aquellos que presentaron LB o EH visible por ultrasonido fueron considerados como casos y se compararon con controles sanos.
Los criterios de inclusión fueron: a) edad entre 20 y 60 años; b) mujeres no embarazadas; c) aceptación para participar en el estudio. Los criterios de exclusión fueron: a) colecistectomía previa; b) lodo biliar por ultrasonido; c) ingesta de alcohol mayor de 40 g/día; d) uso de medicamentos hepa–totóxicos; e) cuadro de colecistitis aguda; f) hepatopatía crónica de etiología viral o metabólica.
Los criterios de eliminación incluyeron aquellos casos en los que la muestra biológica fue insuficiente para los procedimientos analíticos.
Ultrasonido abdominal
El ultrasonido en tiempo real se realizó bajo condiciones de ayuno. Todos los estudios ultrasonográficos fueron evaluados (en dos ocasiones) por el mismo radiólogo (en forma ciega). No hubo discrepancias entre los resultados de la primera y la segunda evaluación, observando concordancia (K = 0.93) entre una y otra evaluaciones.
Definiciones operacionales de LH Y EH
Se definió LB como la presencia de un eco intenso intraluminal que dependía de la gravedad o atenuaba la transmisión (sombra acústica).
El protocolo utilizado para evaluar el patrón de EH por ultrasonido se graduó de la siguiente forma: (0) normal, (1) difuso homogéneo, (2) patrón geográfico: clara diferencia entre el hígado normal y el hígado graso, no confinada a la distribución lobar y sin efecto de masa sobre los vasos hepáticos, (3) focal: sin efecto de masa, sin desplazamiento de vasos, distribución segmentaria o lobar, baja atenuación, patrón en "terremoto" (líneas dispersas de baja atenuación), (4) afección generalizada con una zona normal: pseudotumor (anterior a la vena portal derecha/ fosa de la vesícula biliar, segmento medial del lóbulo izquierdo y porta), (5) patrón en "guante" (digitaciones de alta atenuación de zonas normales rodeadas de un fondo de baja atenuación), simulando metástasis. La intensidad de la infiltración hepática grasa se midió de la siguiente forma: normal; grado 1, atenuación hepática menor que la esplénica; grado 2, diferencia más pronunciada entre hígado y bazo, vasos intrahepáticos no visibles o con atenuación ligeramente menor que la hepática; grado 3, atenuación marcadamente reducida con gran contraste entre el hígado y los vasos intrahepáticos.27
Procedimientos analíticos
Las concentraciones plasmáticas de insulina fueron determinadas por inmunoensayo (MEIA; Abbott Diagnostics), con coeficientes de variación inter e intraanálisis menores a 3 %. Las concentraciones en suero de leptina se determinaron por radioinmunoanálisis usando un estuche comercial (Linco Research, Sto Charles, MO, USA). Los coeficientes de variación inter e intraanálisis fueron menores de 5%. La glucosa plasmática en ayuno se cuantificó por duplicado con un analizador automático, el coeficiente de variación para una determinación única fue de 1.5%
Las concentraciones de colesterol total, C–HDL y triglicé–ridos se determinaron por métodos enzimáticos colorimétricos, usando CHOL, HDL–C plus (segunda generación) y ensayos TG (Roche Diagnostics Co., Indianapolis, IN). Las concentraciones de colesterol asociado a lipoproteínas de baja densidad (C–LDL) se calcularon usando la fórmula de Friedewald.28
Análisis estadístico
Las diferencias entre los promedios de edad, IMC, leptina, e insulina en uno y otro grupos fueron analizadas mediante la prueba de t de Student. Se realizó un análisis de correlación de Pearson para el análisis de asociación entre IMC, leptina e insulina. Las diferencias fueron consideradas estadísticamente significativas cuando el valor de p fue <0.05.
Resultados
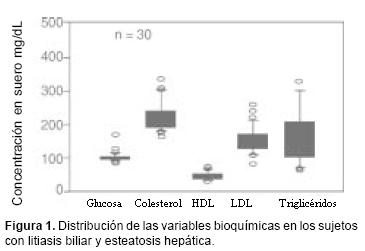
Se estudiaron 317 sujetos, las características clínicas de los mismos se encuentran resumidas en el cuadro I. La edad del grupo control fue significativamente mayor (LB, 52.6 ± 11.6; EH, 49.8 ± 11.1; LB + EH, 51.6 ± 10.5; 57.1 ± 7.4), p< 0.05. El IMC fue mayor en los grupos de EH (28.7 ± 2.8) y LB +EH (29.0 ± 3.8) que en los grupos de LB (27.4 ± 4.3) y controles (51.6 ± 10.5), p< 0.05. El grupo de LB presentó las concentraciones más elevadas de leptina (13.7 ± 8.1) comparado con los otros grupos, la diferencia fue estadísticamente significativa, p < 0.05, (Cuadro II). Mientras que las concentraciones de insulina fueron similares en los cuatro grupos de sujetos, cuadro II. La figura 1 muestra las concentraciones de glucosa 102. 7 ± 15.2 mg/dL, colesterol 225.7 ± 45.1 mg/dL, triglicéridos 184.6 ± 144.2 mg/dL, C–HDL 46.1 ± 12.7 mg/dL, C–LDL 148.5 ± 40.1 mg/dL del grupo de LB + EH. En este mismo grupo se observó una correlación positiva entre el IMC y las concentraciones de leptina, r=0.39, p<0.03 (Figura 2) e insulina, r=0.57, p< 0.0001 (Figura 3).


Discusión
Dado que nuestra hipótesis de trabajo fue que la leptina participa en la patogénesis de la LB y EH, el objetivo de este trabajo fue investigar si existe una relación entre la LB y la EH en donde la leptina juegue un papel importante.
En el presente estudio observamos que las concentraciones de insulina fueron similares en los cuatro grupos de estudio con una tendencia a ser menores en el grupo control, aunque la diferencia no fue estadísticamente significativa. Mientras que las concentraciones de leptina fueron superiores en los grupos de LB y EH.
El papel de la leptina en el desarrollo de LB ha sido descrito inicialmente en población México–americana observando que es un factor de riesgo importante para el desarrollo de LB independientemente del género.29 Este marcador parece no ser más eficaz que el uso de antropometría,30 aunque esto ha sido puesto en duda recientemente en un estudio de Ko et al.31 realizado en mujeres puérperas donde por medio de análisis multivariado ajustado para variables antropométricas, se demuestra el papel predictor de la leptina en el desarrollo de LB. A pesar de estas asociaciones el mecanismo por el que favorece el desarrollo de LB no está bien determinado. Diversos informes en modelos animales han demostrado que ratones resistentes a leptina (mecanismo observado en la mayoría de los sujetos con obesidad) presentan: 1) incremento en el volumen de la vesícula biliar, 2) disminución de la secreción biliar de colesterol, así como de la formación de cristales de colesterol, a pesar de, 3) presentar mayores concentraciones de colesterol en suero que ratones no obesos. Por lo que el vínculo entre obesidad y formación de LB no requiere únicamente de la hipersecreción de colesterol y también puede estar en relación con alteraciones en la motilidad de la vesícula.32–38
Por otro lado, la leptina también tiene un papel muy importante en la fisiopatología de la EHNA. Los informes iniciales mostraron que la administración de leptina en modelos animales ocasiona regresión de las características fenotípicas asociadas a la lipodistrofia congénita generalizada (resistencia a la insulina y EH).39 Este fenómeno indica que la falta de respuesta biológica a la acción de la leptina se encuentra asociada con alteraciones en el metabolismo de la insulina, permitiendo el desarrollo de resistencia a la insulina, piedra angular en la fisiopatológía de la EHNA.24,25,40 Posteriormente estos hallazgos han sido reproducidos en diversos estudios en humanos.41,42 Considerando que la leptina tiene un papel importante en el desarrollo del proceso inflamatorio43 mediado por incremento en la susceptibilidad a estímulos nocivos mediado por liberación de citocromo c, activación de caspasa 3, alteraciones en la transducción de las vías de señalización dependientes del factor nuclear KB,44 y activación de las células estelares, participando así el proceso de fibrosis asociado a EHNA.18,45,46 Sin embargo existen resultados controversiales en relación a la función de la leptina como factor promotor de fibrosis en la EHNA, debido a procesos de desacoplamiento de la cadena respiratoria como efectos protectores en modelos animales,47 y más recientemente en estudios clínicos en humanos la leptina parece no influir en la fibrosis de sujetos con EHNA,48–51 incluso parece ser que ésta no influye en la elevación de enzimas hepáticas en estos sujetos.52 Esto indica que las concentraciones de suero de la leptina determinan la intensidad de la EH53 pero no participan en el proceso inflamatorio, ni en el desarrollo de fibrosis.54,55 En el presente estudio se observó correlación positiva entre las concentraciones de leptina e insulina con el IMC en el grupo de sujetos con LB y EH, lo que indica la importancia de la leptina en estos sujetos.
En conclusión, las concentraciones circulantes de leptina se encuentran elevadas en las enfermedades hepatobiliares (EH y LB) asociadas a la obesidad; sin embargo se requieren más estudios para determinar el mecanismo mediante el cual ocurren estas alteraciones.
Referencias
1. Aguilar–Salinas CA, Vázquez–Chávez C, Gamboa–Marrufo R, García–Soto N, de Jesús Ríos–González J, Holguín R, et al. Obesity, diabetes, hypertension, and tobacco consumption in an urban adult Mexican population. Arch Med Res 2001; 32:446–53. [ Links ]
2. Arroyo P, Loria A, Mendez O. Changes in the household calorie supply during the 1994 economic crisis in Mexico and its implications on the obesity epidemic. Nutr Rev 2004; 62:S163–8. [ Links ]
3. del Río–Navarro BE, Velázquez–Monroy O, Sánchez–Castillo CP, Lara–Esqueda A, Berber A, Fanghanel G, et al. The high prevalence of overweight and obesity in Mexican children. Obes Res 2004; 12:215–223. [ Links ]
4. Villa AR, Escobedo MH, Méndez–Sánchez N. Estimates and trends of obesity prevalence through mortality rates associated of chronic diseases in Mexico. Gac Med Mex 2004; 140(Suppl 2) :S21–S25. [ Links ]
5. Sánchez–Castillo CP. Epidemiología de la obesidad, in Obesidad: epidemiología, fisiopatología, y manifestaciones clínicas, Méndez–Sánchez N, Uribe M, Editors, México. Manual Moderno: 2002. p. 5–31. [ Links ]
6. Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, et al. Prevalence of obesity, diabetes, and obesity–related health risk factors, 2001 Jama 2003; 289:76–79. [ Links ]
7. Méndez–Sánchez N, Sánchez–Castillo CP, Villar AR, Madrigal H, Merino B, García E, et al. Relación entre sobrepeso y obesidad con mortalidad por cirrosis hepática en México. Rev Gastr Mex 2003; 68:176. [ Links ]
8. Méndez–Sánchez N, Sánchez–Castillo CP, Villa AR, Madrigal H, Merino B, García E, et al. The relationship of overweight and obesity to high mortality rates from liver cirrhosis in Mexico. Ann Hepatol 2004; 3:66–71. [ Links ]
9. Tolman KG, Fonseca V, Tan MH, Dalpiaz A. Narrative review: hepatobiliary disease in type 2 diabetes mellitus. Ann Intern Med 2004; 141:946–56. [ Links ]
10. Bennion LJ, Grundy SM. Effects of obesity and caloric intake on biliary lipid metabolism in mano J Clin Invest 1975; 56: 996–1011. [ Links ]
11. StahIberg D, Rudling M, Angelin B, Bjorkhem I, Forsell P, Nilsell K, et al. Hepatic cholesterol metabolism in human obesity. Hepatology 1997;25:1447–50. [ Links ]
12. Lizardi–Cervera J, Motola–Kuba D, Guevara–Gonzalez L. Obesity and its association with cryptogenic cirrhosis and hepatocarcinoma. Gac Med Mex 2004; 140 (Suppl 2):S77–83. [ Links ]
13. Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980; 55:434–438. [ Links ]
14. Ruhl CE, Everhart JE. Determinants of the association of overweight with elevated serum alanine aminotransferase activity in the United States. Gastroenterology 2003; 124:71–79. [ Links ]
15. Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology 2002; 122:1649–1657. [ Links ]
16. Falck–Ytter Y, Younossi ZM, Marchesini G, McCullough AJ. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin Liver Dis 2001;21:17–26. [ Links ]
17. Marceau P, Biron S, Hould FS, Marceau S, Simard S, Thung SN, et al. Liver pathology and the metabolic syndrome X in severe obesity. J Clin Endocrinol Metab 1999; 84: 1513–7. [ Links ]
18. Nakao K, Nakata K, Ohtsubo N, Maeda M, Moriuchi T, Ichikawa T, et al. Association between nonalcoholic fatty liver, markers of obesity, and serum leptin level in young adults. Am J Gastroenterol 2002; 97:1796–1801. [ Links ]
19. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372:425–432. [ Links ]
20. Méndez–Sánchez N, Sánchez–Lara K, Villa AR, Bahena–Aponte J, Chávez Tapia NC, Ramos MH, et al. Adiponectin concentration as protective factor in hepatic steatosis. World J Gastroenterol 2005; in press. [ Links ]
21. Meier U, Gressner AM. Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin, and resistin. Clin Chem 2004; 50:1511–1525. [ Links ]
22. Méndez–Sánchez N, González V, King–Martínez AC, Sánchez H, Uribe M. Plasma leptin and the cholesterol saturation of bile are correlated in obese women after weight loss. J Nutr 2002; 132: 2195–8. [ Links ]
23. Méndez–Sánchez N, Chávez–Tapia N, Uribe M. Effects of leptin on biliary lipids: potential consequences for gallstone formation and therapy in obesity. Curr Drug Targets Immune Endocr Metabol Disord 2005; 5:in press. [ Links ]
24. Méndez–Sánchez N, Chávez– Tapia NC, Uribe M. [Obesity and non–alcoholic steatohepatitis]. Gac Med Mex 2004; 140(Supp12):567–72. [ Links ]
25. Méndez–Sánchez N, Chávez–Tapia NC, Uribe M. An update on non alcoholic fatty liver disease. Rev Invest Clin 2004; 56: 72–82. [ Links ]
26. Li Z, Un H, Yang S, Diehl AM. Murine leptin deficiency alters Kupffer cell production of cytokines that regulate the innate immune system. Gastroenterology 2002; 123:1304–1310. [ Links ]
27. Saadeh S, Younossi ZM, Remer EM, Gramlich T, Ong JP, Hurley M, et al. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology 2002; 123: 745–50. [ Links ]
28. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low–density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972; 18:499–502. [ Links ]
29. Duggirala R, Mitchell BD, Blangero J, Stern MP. Genetic determinants of variation in gallbladder disease in the Mexican–American population. Genet Epidemiol 1999; 16:191–204. [ Links ]
30. Ruhl CE, Everhart JE. Relationship of serum leptin concentration and other measures of adiposity with gallbladder disease. Hepatology 2001; 34:877–83. [ Links ]
31. Ko CW, Beresford SA, Schulte SJ, Matsumoto AM, Lee SP. Incidence, natural history, and risk factors for biliary sludge and stones during pregnancy. Hepatology 2005;41:359–365. [ Links ]
32. Graewin SJ, Lee K, Tran KQ, Goldblatt MI, Svatek CL, Nakeeb A, et al. Leptin–resistant obese mice do not from biliary crystals on a high cholesterol diet. J Surg Res 2003; 114:291. [ Links ]
33. Graewin SJ, Lee KH, Tran KQ Goldblatt MI, Svatek CL, Nakeeb A, et al. Leptin–resistant obese mice do not form biliary crystals on a high cholesterol diet. J Surg Res 2004; 122:145–149. [ Links ]
34. Graewin SJ, Kiely JM, Lee KH, Svatek CL, Nakeeb A, Pitt HA. Nonobese diabetic mice have diminished gallbladder motility and shortened crystal observation time. J Gastrointest Surg 2004; 8: 824–9; discussion 829–30. [ Links ]
35. Graewin SJ, Lee KH, Kiely JM, Svatek CL, Nakeeb A, Pitt HA. Gallbladder myocytes are short and cholecystokinin–resistant in obese diabetic mice. Surgery 2004; 136:431–436. [ Links ]
36. Tran KQ Goldblatt MI, Swartz–Basile DA, Svatek C, Nakeeb A, Pitt HA. Diabetes and hyperlipidemia correlate with gallbladder contractility in leptin–related murine obesity. J Gastrointest Surg 2003; 7:857–62; discussion 863. [ Links ]
37. Tran KQ, Graewin SJ, Swartz–Basile DA, Nakeeb A, Svatek CL, Pitt HA. Leptin–resistant obese mice have paradoxically low biliary cholesterol saturation. Surgery 2003; 134: 372–7. [ Links ]
38. Tran KQ, Swartz–Basile DA, Nakeeb A, Pitt HA. Gallbladder motility in agouti–yellow and leptin–resistant obese mice. J Surg Res 2003; 113: 56–61. [ Links ]
39. Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 1999; 401: 73–6. [ Links ]
40. Un HZ, Yang SQ, Chuekaree C, Kuhajda F, Ronnet G, Diehl AM. Metformin reverses fatty liver disease in obese, leptin–deficient mice. Nat Med 2000; 6:998–1003. [ Links ]
41. Tobe K, Ogura T, Tsukamoto C, Imai A, Matsuura K, Iwasaki Y, et al. Relationship between serum leptin and fatty liver in Japanese male adolescent university students. Am J Gastroenterol 1999; 94:3328–35. [ Links ]
42. Giannini E, Botta F, Cataldi A, Teneoni GL, Ceppa P, Barreea T, et al. Leptin levels in nonalcoholic steatohepatitis and chronic hepatitis C. Hepatogastro–enterology 1999; 46:2422–2425. [ Links ]
43. Uygun A, Kadayifci A, Yesilova Z, Erdil A, Yaman H, Saka M, et al. Serum leptin levels in patients with nonalcoholic steatohepatitis. Am J Gastroenterol 2000; 95:3584–3589. [ Links ]
44. Yang S, Lin H, DiehI AM. Fatty liver vulnerability to endotoxin–induced damage despite NF–kappa B induction and inhibited caspase 3 activation. Am J Physiol Gastrointest Liver Physiol 2001; 281 :G382–92. [ Links ]
45. Leclercq lA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol 2002; 37:206–13. [ Links ]
46. Tomita K, Azuma T, Kitamura N, Tamiya G, Ando S, Nagata H, et al. Leptin deficiency enhances sensitivity of rats to alcoholic steatohepatitis through suppression of metallothionein. Am J Physiol Gastrointest Liver Physiol 2004; 287:G1078–G1085. [ Links ]
47. Baffy G, Zhang CY, Glickman JN, Lowell BB. Obesity–related fatty liver is unchanged in mice deficient for mitochondrial uncoupling protein 2. Hepatology 2002; 35:753–761. [ Links ]
48. Angulo P, Alba LM, Petrovic LM, Adams LA, Lindor KD, Jensen MD. Leptin, insulin resistance, and liver fibrosis in human nonalcoholic fatty liver disease. J Hepatol 2004; 41:943–949. [ Links ]
49. Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF–alpha or adiponectin? Hepatology 2004; 40:46–54. [ Links ]
50. Chalasani N, Crabb DW, Cummings OW, Kwo PY, Asghar A, Pandya PK, et al. Does leptin play a role in the pathogenesis of human nonalcoholic steatohepatitis? Am J Gastroenterol 2003; 98:2771–2776. [ Links ]
51. Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 2002; 109:1345–50. [ Links ]
52. Liangpunsakul S, Chalasani N. Relationship between unexplained elevations in alanine aminotransferase and serum leptin in U.S. adults: results from the Third National Health and Nutrition Examination Survey (NHANES III). J Clin Gastroenterol 2004; 38:891–897. [ Links ]
53. Gunel N, Coskun U, Toruner FB, Sancak B, Yilmaz E, Cengiz O, et al. Serum leptin levels are associated with tamoxifen–induced hepatic steatosis. Curr Med Res Opin 2003; 19:47–50. [ Links ]
54. Serin E, Ozer B, Gumurdulu Y, Kayaselcuk F, Kul K, Boyacioglu S. Serum leptin level can be a negative marker of hepatocyte damage in nonalcoholic fatty liver. J Gastroenterol 2003; 38:471–476. [ Links ]
55. Chitturi S, Farrell G, Frost L, Kriketos A, Lin R, Fung C, et al. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology 2002; 36:403–409. [ Links ]

