











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkThis paper is dedicated to Prof. Jorge Ibañez and Prof. Yunny Meas for their experimental and scientific works about the chemical education and environmental electrochemistry, for their scientific contributions related to the topics of the electrochemical technologies as well as for the efforts to disseminate the electrochemistry in different Latin American countries.
Introduction
Electrochemical oxidation (EO) is one of the most effective and widely studied electrochemical technologies for the remediation of organic pollutants from water/wastewater [1-5]. It is the simplest and the most popular of the electrochemical advanced oxidation processes (EAOPs) [1,6,7]. In EO, the organic compounds contained in contaminated solutions or wastewater are oxidized by direct charge transfer on the anode surface (M) or destroyed by the physisorbed hydroxyl radicals (M(●OH)) generated as an intermediate of O2 evolution from the water oxidation reaction at high current densities on the anodal surface according to Eq. 1 [1,7,8]:
The types of electrocatalytic materials used to determine the amount and nature of the generated M(●OH). While some anode materials such as Pt, carbon and dimensional stable anodes (active anodes) strongly interact with the M(●OH) produced to form chemisorbed oxygen or superoxide, which is a weaker oxidant compared to free-●OH, they only allow conversion of organic compounds into stable intermediates with limited mineralization [1,9,10]. Other materials, such as Sn and Pb-doped oxides, substoichiometric TiO2 and boron-doped diamond (BDD) [11-14], all of them considered non-active anodes, and with high O2 over-potentials, weakly interact with the M(●OH), promoting the degradation and mineralization of organic pollutants and their aromatic intermediates until their total conversion to CO2 [15-18]. Previous studies have demonstrated the potential of EO with both types of anodes for degrading different classes of organic pollutants, and though both types of anodes usually lead to similar of oxidation products, the non-active variety enables greater and faster mineralization [2,19-23]. In addition, EO using flow reactors exhibits a number of advantages, including easy automation and scale-up, as well as the ability to perform continuous operation and treat large volumes of wastewater with a small electrode area compared to non-flow reactors [1].
Wastewater containing synthetic textile dyes is one of the major industrial effluents posing a global environmental threat because these compounds are produced in large volumes and concentrations [16-18,24]. The industrialization has significantly increased the use of coloring chemicals such as dyes, with over 10,000 different types of dyes and pigments manufactured worldwide [25-28]. A number of industries (textile, leather, food, cosmetic, pharmaceutical, paper etc.) use different varieties of dyestuffs, although the textile industry is the major consumer of dyestuffs and pigments, accounting for the largest amount of dye-contaminated effluents production and dyeing processes [22,24]. The occurrence of dyestuffs in water systems/environment is a serious concern for regulatory agencies due to their potentially toxic, teratogenic, mutagenic and carcinogenic effects, parent compounds and oxidation by-products such as aromatic amines and anilines [6,29,30]. Most synthetic dyestuffs exhibit complex structures that provide them with high chemical, photolytic and biological stability, resulting in their persistence and accumulation in water bodies [30,31]. The colors used in textile and pigment industries are achieved by mixing two or more different dye compounds with optimum reactivity, dispersity and diffusion behaviour via a process known as “trichromy formulation” [32]. As such, effluents and wastewater from textile and other coloring-related industries contain not only a single dye compound, but a trichromy of dyestuffs made up of several dyes with different structures, persistence, and toxicity [32]. The presence of these pollutants in the environment reduces dissolved oxygen and light penetration, especially in aquatic systems, and if accumulated in large amounts, may result in esthetic pollution [29,32]. In addition to the dyes, a trichromy contains surfactants, sulfates, chlorides, hydroxides and heavy metals, depending on the textile substrate being processed [32]. The multiple components and complexity of textile-dyeing plant effluents make them extremely difficult to treat by conventional methods employed in municipal or sewage treatment plants [1].
Advanced oxidation processes (AOPs) based on the generation of powerful oxidizing agents such as ●OH, which allow quick and non-selective degradation of organic contaminants into biodegradable by-products (such as, carboxylic acids [33-37]) or complete conversion to CO2, have been investigated for the treatment of several single or multi-component synthetic or real effluents containing dyestuffs [1,15,18,22]. Indeed, there are numerous studies and a few review papers on electrochemical technologies, including EO, as effective techniques for removing dyestuffs from wastewater [22]. However, most focused on a single-component solution with limited consideration of effluents containing multiple components or trichromy dyestuffs [32].
In this paper, we investigate the capacity of EO to remove colour and dissolved organic matter from real wastewater obtained from the trichromy of dyes used in fabric-dyeing processes in a parallel plate continuous single flow reactor. The effect of applied current density and anode material (i.e., Ti/Pt or Ti/Pt-Sb-Sn) on colour removal efficiency, COD, turbidity and DO during the treatment of single and trichromy dye solutions was examined. The data obtained were used to estimate electrical energy consumption and cost.
Experimental
Chemicals/materials
The real textile effluents used in this study were provided by the Laboratory of Textile Chemical Processes of the Federal University of Rio Grande do Norte, Natal Brazil. Wastewater containing a single sample of Remazol Yellow 3RS (RY 3RS), Remazol Red RR Gran (RR-RR Gran) and Navy-Blue CL-R (NB CL-R) was collected after a cotton-dyeing process with each of the dyes using a common textile process used by Brazilian industries. The structure and some characteristics of these dyes are shown in Table 1. A trichromy textile effluent was generated after using a mix of three dyes (Trichromy): RY 3RS, RR-RR Gran and NB CLR in the textile fabric. The dyeing process was performed in a BMA-B MATHIS device with eight 260 mL cylinders in a solution containing 1 % wt of dye (single dye process) or 0.3% wt of each dye (trichromy of dyes), and auxiliary agents consisting of 20 g L-1 NaCl, 20 g L-1 Na2CO3, and 3 g L-1 NaOH at 60°C. The electrochemical experiments were performed directly with these effluents because of to their good electrical conductivity due to the presence of the auxiliary agents that act as electrolytes.
Table 1 The structure and characteristics of dyes.
| Commercial name Color index name | Remazol Yellow 3RS Reactive Yellow 176 | Navy Blue CL-R Reactive Blue 19 | Remazol RED RR Gran Reactive Red 198 |
| Chemical structure |

|
 |
 |
| Chemical class | Monoazo | Anthraquinone | Monoazo |
| Molecular formula | C29H21ClN8Na4O16S5 | C22H16N2Na2O11S3 | C27H18ClN7Na4O16S5 |
| Color index number | - | 61200 | 18221 |
| λmax (nm) | 420 | 590 | 515 |
| MW (g/mol) | 1025.26 | 626.55 | 984.21 |
Electrolytic flow reactor and experimental procedures
The EO of the textile effluents was conducted in a single flow cell with a reaction compartment capacity of 110 cm3 (Fig. 1). The reactor consists of two parallel disc plates as anodes. The anodes used were a thin film of platinum (Pt) or mixed Pt-Sn-Sb deposited on Ti ((Ti/Pt) or Ti/Pt-Sn-Sb)) with a 63.6 cm2 geometrical area, while a Ti plate was used as cathode. Both materials were provided by Industrie De Nora Elettrodi (Milan, Italy). All experiments were performed with MINIPA MPL - 3305 triple power supply at constant current. Electrolytic trials were conducted with 2 L of the wastewater stored in a thermo-regulated reservoir and circulated through the cell by means of a peristatic pump at a flow rate of 160 dm3 h-1 (Fig. 1). Samples were withdrawn at predetermined time intervals during the electrochemical treatment for analysis of colour and COD removal, turbidity and DO, and these parameters were used to assess the efficiency of the process.

Instruments and analytical procedures
The colour removal of the treated solution was followed by absorbance spectrum measurement of the dyes at 620 nm, 509 nm, 435 nm, 509 nm for NB CL-R, RR-RR Gran, RY 3RS, and trichromy, respectively, in a SPECORD®210 Plus spectrophotometer (Analytikjena, Germany) at room temperature. The decolourization efficiency or percentage of colour removal as a function of electrolysis time during dye solution treatment was calculated from the absorbance at time zero (A 0 ) and after electrolysis time t (A t ), according to the following relation [24]:
where ABS 0 M and ABS t M are the average absorbance before electrolysis and after electrolysis time t, respectively, at the maximum visible wavelength ((max = 620 nm, 509 nm, 435 nm, 509 nm for NB CL-R, RR RR Gran, RY 3RS and trichromy respectively).
Removal of the organic matter content of the treated effluents was assessed by monitoring COD using pre-dosage HANNA® vials containing 2 mL of sample. Samples to measure COD were digested in an HI 839800 thermoreactor (HANNA Instruments) at 150 °C for 150 min, allowed to cool to room temperature and analyzed in a photometer (Hanna HI table 83099) [38]. The progress of dissolved O2 concentrations and solution turbidity during electrochemical treatment were measured with an INSTRUTHERM MO-900 oximeter and INSTRUTHERM TD-300 turbidity meter, respectively. The cell voltage displayed on the MINIPA MPL-3305 triple power supply during electrolysis was recorded as a function of time and energy consumption per volume of treated effluent was estimated using the relation in Equation 3 [24]:
where V is the cell voltage (v), A the applied current (A), t electrolysis time (h) and Vs the volume of the treated wastewater (m3).
Results and discussion
Behavior of the dye absorbance spectra
The behaviour of the absorbance bands (λmax = 616 nm) obtained for NB CL-R during EO treatment with the Ti/Pt and Ti/Pt-Sn-Sb anodes at 30 and 60 mA cm-2 is shown in Fig. 2(a-d). The intensity of all the peaks in the UV-Vis regions decreased progressively to zero with electrolysis for 240 min in all cases, confirming the effectiveness of EO under the study conditions in degrading organic molecules and decolorizing dye solutions. Faster absorption band decrease was achieved at the highest current density (i.e., 60 mA cm-2, Fig. 2(b) and 2(d) compared to the lowest (30 mA cm-2, Fig. 2(a) and 2(c). The complete disappearance of the NB CL-R absorbance band was observed after 120 min of electrolysis at 60 mA cm-2 with the Ti/Pt anode, whereas 180 min was required to totally eliminate the representative color band at 30 mA cm-2, using the same anode [42-44]. Moreover, a faster decrease in the absorbance bands of this dye was achieved with the Ti/Pt-Sn-Sb anode compared to Ti/Pt at 30 and 60 mA cm-2 as well as all the electrolysis times studied, indicating high oxidation potential of Ti/Pt-Sn-Sb for degrading dyes in solution.
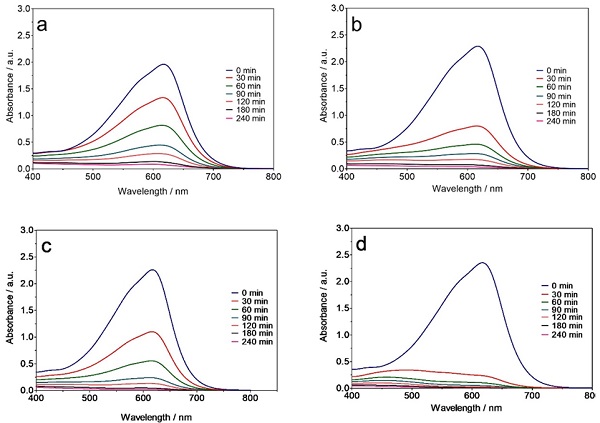
Fig. 2 Behaviour of the NB CL-R absorbance band (λmax = 590 nm), as a function of time, during EO of 2 L of effluent with (a, b) Ti/Pt and (c, d) Ti/Pt-Sn-Sb applying (a, c) 30 and (b, d) 60 mA cm-2.
Similar trends for decreasing intensity of peaks in the UV-vis region were observed for other dyes (Figures 3(a-d) and 4(a-d)) and the trichromy (Fig. 5(a-d)) using either Ti/Pt or Ti/Pt-Sn-Sb. Moreover, a faster decrease in the absorbance bands of the dyes was achieved with the Ti/Pt-Sn-Sb anode compared to Ti/Pt at 30 and 60 mA cm-2 as well as all the electrolysis times studied, indicating high oxidation potential of Ti/Pt-Sn-Sb for degrading dyes in solution. Faster absorption band decrease was achieved at the highest current density (i.e., 60 mA cm-2) compared to the lowest (30 mA cm-2) regardless of dye solution treated and electrode used. This behaviour can be explained by the efficient production of oxidants, especially M(●OH) from water oxidation (Eq. 1), which can quickly oxidize the colourants in the solution, thereby diminishing the intensity of the absorbance bands [6,39-41].
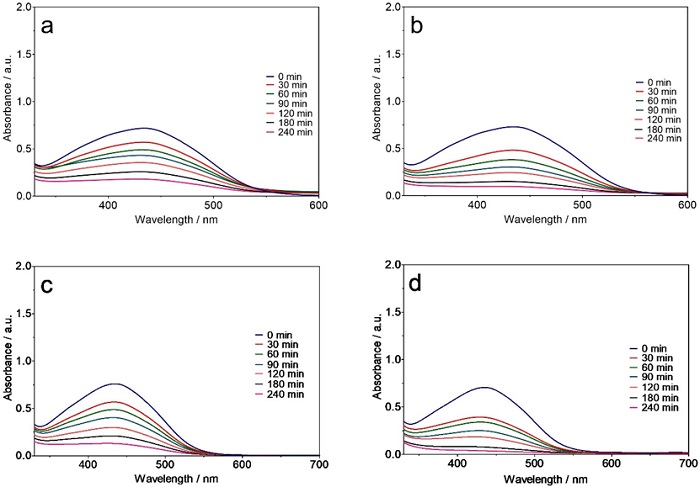
Fig. 3 Behaviour of the RY 3RS absorbance band (λmax = 420 nm), as a function of electrolysis time, during EO of 2 L of effluent with (a, b) Ti/Pt and (c, d) Ti/Pt-Sn-Sb applying (a, c) 30 and (b, d) 60 mA cm-2.
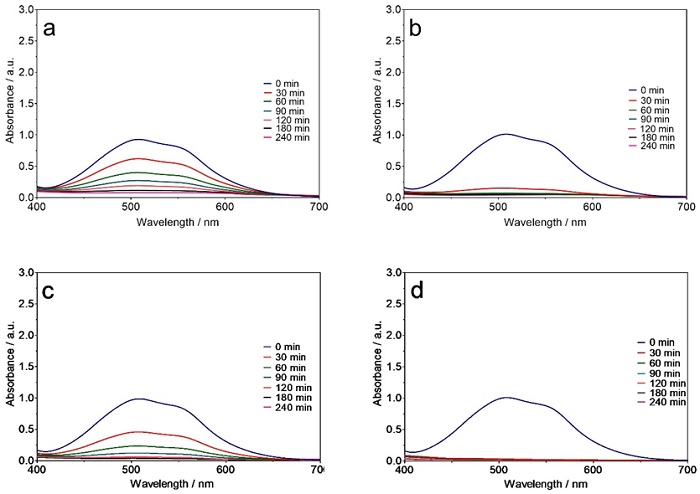
Fig. 4 Behaviour of the RR-RR Gran absorbance band (λmax = 515 nm) as a function of electrolysis time, during EO of 2 L of effluent with (a, b) Ti/Pt and (c, d) Ti/Pt-Sn-Sb applying (a, c) 30 and (b, d) 60 mA cm-2.
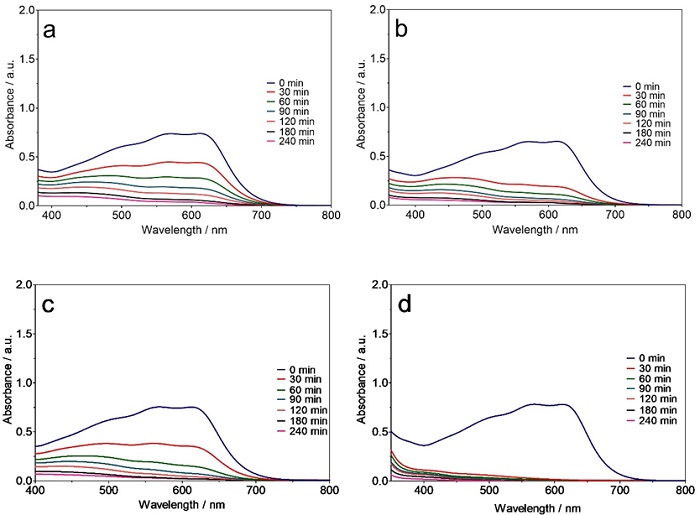
Fig. 5 Behaviour of the trichromy effluent absorbance band (λmax = 568 nm), as a function of time, during EO of 2 L of effluent with (a, b) Ti/Pt and (c, d) Ti/Pt-Sn-Sb by applying (a, c) 30 and (b, d) 60 mA cm-2.
The rapid decrease in the absorbance bands of the dyes and limited shift in the maximal wavelengths suggested that decolourization of the treated solution involves the oxidation of the dye into other organic compounds without transforming them into other coloured compounds that can affect or shift the maximal band wavelength [44]. Previous studies have demonstrated that the oxidation of dyes usually occurs via the formation of cyclic and non-cyclic intermediates (aromatic, aliphatic carboxylic acids and carbon dioxide), after the cleavage or fragmentation of their structure in the chromophore group [44-46].
Fig. 6 shows the percentage of color removal for NB CL-R, RR RR Gran, RY 3RS and trichromy effluents, as a function of electrolysis time, obtained during the EO process. As can be seen in Fig 6, complete decolourization of the dye solutions was achieved irrespective of applied current density, except for RY 3RS with the Ti/Pt anode, which could only achieve 75 % and 67 % colour removal in 240 min of electrolysis at 30 and 60 mA cm-2, respectively. This behaviour showed that dye molecules exhibit different reactivity rates with the oxidants generated and that oxidation efficiency has a strong relationship with the chemical structure of the treated pollutant [46,47]. Slower decolourization of the RY 3RS dye solution (82.8% and 94.8% after 240 min of electrolysis at 30 and 60 mA cm-2, respectively) was also observed during electrolysis with the Ti/Pt-Sn-Sb anode compared to the other dyes and trichromy solution, at all current density values used, confirming the relatively higher resistance of RY 3RS to oxidation when compared to the other dye solutions studied [47]. Similar behaviour has been reported for the electrochemical degradation of pharmaceutical beta blockers (atenolol, metoprolol and propranolol), with the kinetic rate of pharmaceutical oxidation depending on the number of benzoic moieties in the aromatic part of their chemical structures [48].
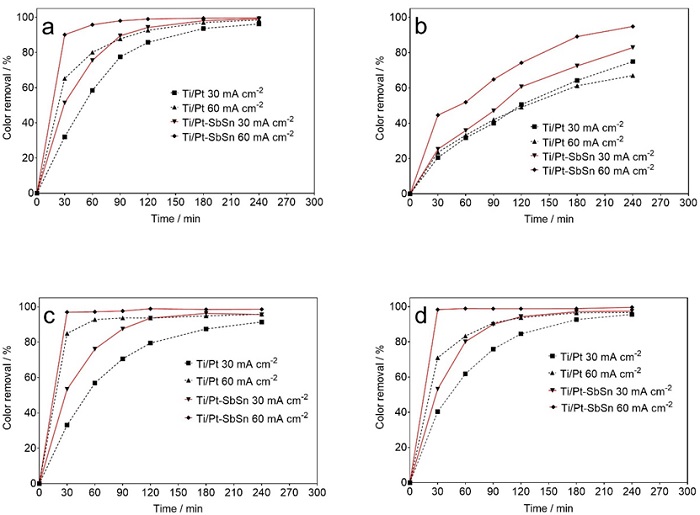
Fig. 6 Percentage colour removal vs electrolysis time for the EO treatment of 2 L solution of: (a) NB CL-R, (b) RY 3RS, (c) RR-RR Gran and (d) trichromy at (■, ▼) 30 and (▲, ♦) 60 mA cm-2 using (■, ♦) Ti/Pt and (▼, ▲) Ti/Pt-Sn-Sb anode.
Colour removal efficiency was strongly influenced by the applied current density during the EO process [32,38,48]. Faster and higher decolourization efficiency was observed at 60 mA cm-2 compared to 30 mA cm-2 for both anode materials during the treatment of all the dye solutions, which is expected based on the higher M(●OH) production rate from water oxidation on the surface at higher current density [1,15]. Thus, complete decolourization was achieved after 180, 120 and 15 min for the EO treatment of NB-CL-R, RR-RR Gran and trichromy solutions, respectively, at 60 mA cm-2 using a Ti/Pt-Sb-Sn anode; whereas 100 %, 96 % and 97 % decolourization was attained after 240 min of NB-CL-R, RR-RR Gran and trichromy solutions, respectively, at 30 mA cm-2 using the same anode. A similar trend was observed with Ti/Pt except in the case of RY 3RS, where almost the same colour removal was obtained at both current densities up to 180 min of electrolysis. The influence of the anode material on the decolorization of the dye solutions is depicted in Fig. 4 when electrolyzed at either current density. The use of a Ti/Pt-Sn-Sb anode showed remarkable colour removal acceleration at both current densities compared to the Ti/Pt anode. A high O2 over-potential anode, such as Ti/Pt-Sn-Sb, is a non-active electrode with far greater ability to produce active radicals (i.e. M(●OH)) adsorbed weakly compared to a low oxidation power anode such as Ti/Pt [6,24]. Ti/Pt(●OH) is generated in a smaller amount on the Ti/Pt surface because it is further oxidized to chemisorbed oxygen (Eq. 4), which has low oxidation power for the degradation of organic pollutants [1,24].
Degradation of dye solutions
The degradation rates measured under different experimental conditions could be conveniently compared in terms of the observed pseudo-first-order kinetic constants. The estimated kinetic parameters for NB CL-R, RY 3RS, RR-RR Gran and trichromy are summarized in Table 2. In general, the rate of degradation of the dyes and trichromy increased with rising applied current density (from 30 to 60 mA cm-2,) regardless of the type of electrocatalytic material used. However, the increase in the degradation rate was accentuated when using the Ti/Pt-SbSn anode due to its greater capacity to produce oxidants that contribute to the degradation of the dyes, as previously mentioned. Furthermore, it was observed that the degradation of the RR-RR Gran dye at 60 mA cm-2 on the Ti/Pt and Ti/Pt-SbSn anode occurred through two steps, being one of them, a fast step, and the other, a slow step. However, it was only possible to determine the rate constant of the fast step for the Ti/Pt anode (0.0438 min-1), since in the Ti/Pt-SbSn anode the degradation was instantaneous in the range of 0 to 30 minutes. On the other hand, the slow step occurred with kobs of 0.0029 min-1 and 0.0039 min-1 for the Ti/Pt and Ti/Pt-SbSn anode, respectively. The degradation of NB CL-R at 60 mA cm-2 with the Ti/Pt-SbSn anode also occurred in two steps (one fast and the other slow). The same degradation behaviour was observed for trichromy at 60 mA cm-2 with Ti/Pt anode and both current densities with Ti/Pt-Sn-Sb anode. The fast step is associated with the breaking or fragmentation of the chromophore group structure and the slow step with the degradation of cyclic and non-cyclic intermediates.
Table 1 The calculated kinetic parameters for the pseudo-first-order models for the removal of NB CL-R, RY 3RS, RR-RR Gran and trichromy by Ti/Pt and Ti/Pt-SbSn anodes at 30 and 60 mA cm-2.
| kobs/min-1 | ||||
| Ti/Pt | Ti/Pt-Sn-Sb | |||
| 30 mA cm-2 | 60 mA cm-2 | 30 mA cm-2 | 60 mA cm-2 | |
| RY 3RS | 0.0056 | 0.0081 | 0.0072 | 0.012 |
| RR-RR Gran | 0.0103 | 0.0438a/0.0029b | 0.0227a | 0.0039b |
| NB CL-R | 0.0141 | 0.0166 | 0.0202 | 0.0361a/0.0075b |
| Trichromy | 0.0130 | 0.0221a /0.0069b | 0.0241a /0.0069b | 0.0757a |
afirst step of pseudo-first order kinetics and bsecond step of pseudo-first order kinetics.
Elimination of the dye solutions assessed from COD removal after 240 min of EO treatment with either electrode at 30 and 60 mA cm-2 is shown in Fig. 7. Different behaviour was observed for both electrodes in different dye solutions, in terms of COD removal. For example, COD remains constant or increases after the EO treatment of NB CL-R with Ti/Pt-Sn-Sb at 30 mA cm-2, RY 3RS with Ti/Pt-Sn-Sb at both current densities and RR-RR Gran with either anode at both current densities. This phenomenon is attributed to the formation of polymer organic compounds on the anode surface via electrolysis, which avoids further oxidation of the byproducts by the oxidants generated on the anodic surface [46]. In particular, organic molecules with phenol and its derivatives as their major primary by-products exhibit a greater trend to forming polymer films on the anode surface [15,46]. Indeed, the formation of phenolic polymer film on the anode surface has been reported for the EO degradation of phenol and its derivatives as well as other organic compounds such as paracetamol whose oxidation involves the formation of phenol [3,7]. This behaviour is peculiar to “active anodes” such as Ti/Pt and carbon materials, and on rare occasions with low oxidation potential, “non-active” anodes such as substoichiometric TiO2 [1,3]. By contrast, significant COD removal (85 %) was observed for NB CL-R treatment using Ti/Pt at 30 mA cm-2 (Fig. 7(a)) after 240 min of electrolysis. However, poor COD removal was achieved when current density was increased to 60 mA cm-2, which can be attributed to loss of oxidation potential due to polymer film formation on the anode surface and enhanced parasitic reactions, such as recombination of chemisorbed oxygen [42-45]. Different behaviour was observed during EO of RY 3RS (Fig. 7(b)) and trichromy (Fig. 7(d)) solutions with Ti/Pt electrodes. In this case, COD removal increased when current density was rose from 30 to 60 mA cm-2 which is expected from the enhanced generation of oxidants at higher current density. Electrolysis of the trichromy solution with Ti/Pt-Sn-Sb (Fig. 7(d)) showed similar behaviour with better COD removal at high current density. Higher COD removal was obtained with the Ti/Pt-Sn-Sb anode compared to Ti/Pt during the EO of the trichromy solution at both current densities, in line with the higher M(•OH) production on the surface of the former. The contrast behaviour of these electrodes in different treated solutions supports the earlier hypothesis that EO degradation of organic pollutants depends on their chemical structures [32].

Dissolved oxygen and turbidity
Dissolved (DO) is the amount of molecular oxygen (O2) (mg L-1 or percent saturation) dissolved in water at a given temperature and pressure. The discharge of industrial effluents and sewage as well as high temperatures may significantly reduce DO levels in wastewater owing to lower oxygen solubility in water [6,24,30,49,50]. As shown in Table 3, an increase in DO concentration was observed after the EO treatment, regardless of electrode material or dye solution treated. This is expected because an oxygen evolution reaction is favored, increasing DO in the treated solution [6]. Furthermore, higher DO concentrations were achieved with lower COD removal at both current densities when the Ti/Pt anode was used, irrespective of the dye solution studied. For example, at 30 mA cm-2, DO concentrations of 32.96, 34.44 and 63.38 mg L-1 were achieved with COD removal of 87, 13.93 and 12.61 % for NB CL-R, trichromy and RY 3RS, respectively. Similar behavior was observed during dye solution treatment with the Ti/Pt-Sn-Sb anode at DO concentrations of 60.56 and 32.7 mg L-1 obtained for 0.41 and 54.47 % COD removal at 30 mA cm-2 for the RR-RR Gran and trichromy dye solutions, respectively. Moreover, 61.66 and 50.84 mg L-1 of DO was achieved at 7.45 and 48.07 % of COD removal for RR-RR Gran and NB CL-R 60 mA cm-2.
In all the trials conducted, the treated effluent contained high DO values, which is desirable in natural water systems. However, these high-DO effluents are detrimental in water flowing through iron and steel pipes, increasing corrosion [51]. The presence of DO in wastewater is often desirable because it prevents the formation of substances with unpleasant odors that compromise various water uses, such as drinking water. Furthermore, the DO concentration of effluents discharged directly into aquatic compartments should be controlled because most aquatic organisms, especially fish, require DO concentrations between 10 % and 60 % saturation to survive, depending on the species and other water characteristics [30,52,53]. Thus, based on the experimental DO values obtained in this study, direct discharge of the electrochemical effluent into aquatic habitats seems inadvisable. However, when the volume and characteristics of the water bodies are taken into consideration, DO values may be insignificant after effluent dilution.
Table 3 DO and COD removal efficiencies after 240 min of electrolysis using Ti/Pt and Ti/Pt-Sn-Sb.
| Ti/Pt | |||||
| J = 30 mA cm-2 | J = 60 mA cm-2 | ||||
| Dye | DOinitial | COD removal | DO | COD removal | DO |
| NB CL-R | 22.7 | 87.14 | 32.96 | 11.49 | 51.06 |
| RY 3RS | 8.5 | 12.61 | 63.38 | 20.68 | 58.48 |
| RR RR Gran | 8.27 | 0 | 46.42 | 0 | 43.3 |
| trichromy | 6.63 | 13.93 | 34.44 | 30.03 | 31.72 |
| Ti/Pt-Sn-Sb | |||||
| J = 30 mA cm-2 | J = 60 mA cm-2 | ||||
| Dye | DOinitial | COD removal | DO | COD removal | DO |
| NB CL-R | 22.7 | 0 | 32.88 | 48.07 | 50.84 |
| RY 3RS | 8.5 | 0 | 65.62 | 0 | 64.84 |
| RR RR Gran | 8.27 | 0.41 | 60.56 | 7.45 | 61.66 |
| trichromy | 6.63 | 54.74 | 32.7 | 74.48 | 60.4 |
T = 25 ºC; DO: mg/L; COD removal: %.
Effluent turbidity after 240 min of EO is presented in Table 4. Turbidity is the measurement of the reduced intensity of a light beam passing through solution/aqueous media due to the presence of suspended solids, including inorganic particles (sand, sediment, clay) and organic wastes such as algae and bacteria, plankton, etc. High turbidity inhibits photosynthesis in algae and other aquatic vegetation, and may affect the biodiversity of aquatic biological communities [54]. In addition to its color effects, the presence of dye molecules dispersed in water systems increases the turbidity of the aquatic environment [54]. As shown in Table 4, lower turbidity values were obtained with increasing dye removal efficiency, especially for the treatment of NB CL-R and RR-RR Gran dyes with the Ti/Pt anode and trichromy dye solution using the Ti/Pt-Sn-Sb electrode, indicating that eliminating these dyes enhances light movement within the solution. For instance, turbidity values of 14.36 and 4.5 NTU were obtained for NB CL-R treatment after 96.19 % and 99 % decolorization using Ti/Pt at 30 and 60 mA cm-2. Thus, the EO process can contribute to the elimination of suspensions or macromolecules to obtain clean water [24,30,54].
Table 4 Turbidity and color removal after 240 min of electrolysis using Ti/Pt and Ti/Pt-Sn-Sb.
| Ti/Pt | ||||
| J = 30 mA cm-2 | J = 60 mA cm-2 | |||
| Dye | Colour removal | Turbidity | Colour removal | Turbidity |
| NB CL-R | 96.19 | 14.36 | 98.55 | 4.5 |
| RY 3RS | 74.97 | 11.56 | 67.03 | 5.64 |
| RR RR Gran | 91.40 | 16.67 | 95.77 | 11.35 |
| trichromy | 95.60 | 3.9 | 96.78 | 4.63 |
| Ti/Pt-Sn-Sb | ||||
| J = 30 mA cm-2 | J = 60 mA cm-2 | |||
| Dye | Colour removal | Turbidity | Colour removal | Turbidity |
| NB CL-R | 99.19 | 5.91 | 99.60 | 7.72 |
| RY 3RS | 82.88 | 9.67 | 94.84 | 10.39 |
| RR RR Gran | 95.58 | 8.75 | 98.56 | 18.7 |
| trichromy | 97.51 | 12.27 | 99.65 | 2.19 |
T = 25 ºC; Turbidity: NTU; color removal: %.
Cell potential, energy consumption (CE) and cost
Fig. 8 shows the behaviour of cell potentials as a function of electrolysis time obtained for the EO treatment of the dyes. The cell potential was relatively stable for the Ti/Pt-Sn-Sb electrode in all the treated dye solutions and approximately the same cell voltage was displayed in all conditions. This implies that the conductivity of both the electrolyte (in this case the dyeing solutions) and anodes remains largely unchanged during EO processes. By contrast, there was a slight decrease in cell voltage during EO treatment of all the dye solutions with the Ti/Pt anode in the first 60 min, indicating a slight change in the conductivity of the electrolytic system. As expected, high cell voltage was displayed at 60 mA cm-2 compared to 30 mA cm-2 with either electrode, in all treated dye solutions. In addition, higher cell voltage was observed with the Ti/Pt anode compared to the Ti/Pt-Sn-Sb electrode, regardless of the solution and current density. This is attributed to the higher electric conductivity of the latter when compared to the former.
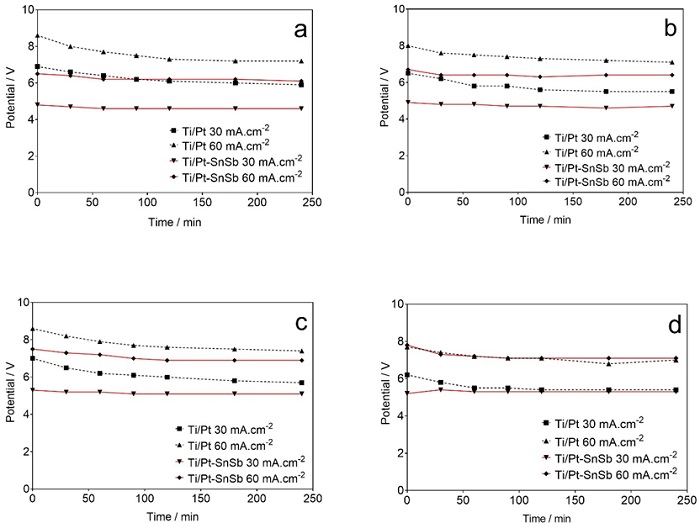
Fig. 8 Variation of cell potential vs electrolysis time for the EO treatment of: (a) NB CL-R, (b) RY 3RS, (c) RR RR Gran and (d) trichromy at (■, ▼) 30 and (▲, ♦) 60 mA cm-2 using (■, ♦) Ti/Pt and (▼, ▲) the Ti/Pt-Sn-Sb anode.
Energy consumption per unit volume of the treated solution and cost, estimated from Equation (3) for all trials after 240 min is exhibited in Table 5. This parameter is important in determining the efficiency of the process, scale-up and adaptation of the technology for commercial applications [1,24]. As illustrated in the Table 5, for all the treated dye solutions, similar energy consumption was obtained for each electrode at a given applied current density. For instance, energy consumption for the EO treatment of any of the dye solutions with the Ti/Pt and Ti/Pt-Sn-Sb anode at 30 and 60 mA cm-2 was 42.75 ± 2 and 108.05 ± 1.65 kWh m-3; and 37.43 ± 2.47 and 100.7 ± 7.22 kWh m-3, respectively. This is promising for the design and scale-up of EO processes for commercial wastewater treatment, since wide varieties of organic pollutants can be treated using a single treatment plant. Moreover, lower energy requirements were observed at low current density as well as in treatment with Ti/Pt-Sn-Sb, regardless of the treated solution, owing to the lower cell voltage and higher conductivity of the Ti/Pt-Sn-Sb anode.
Table 5 Energy consumption (EC) per unit volume of treated effluent during EO treatment of NB CL-R, RY 3RS, RR RR Gran and trichromy by applying 30 and 60 mA cm-2 with Ti/Pt and Ti/Pt-Sn-Sb electrodes.
| Ti/Pt | ||||
| J = 30 mA cm-2 | J = 60 mA cm-2 | |||
| Dye | EC (kW h m-3) | Cost (R$) | EC (kW h m-3) | Cost (R$) |
| NB CL-R | 44.84 | 15.72 | 107.45 | 38.38 |
| RY 3RS | 41.8 | 14.66 | 107.92 | 37.84 |
| RR RR Gran | 43.32 | 15.19 | 110.43 | 39.44 |
| trichromy | 41.04 | 14.39 | 106.4 | 37.31 |
| Ti/Pt-SnSb | ||||
| J = 30 mA cm-2 | J = 60 mA cm-2 | |||
| Dye | EC (kW h m-3) | Cost (R$) | EC (kW h m-3) | Cost (R$) |
| NB CL-R | 34.96 | 12.26 | 92.72 | 32.51 |
| RY 3RS | 35.72 | 12.52 | 97.28 | 34.11 |
| RR RR Gran | 38.76 | 13.59 | 104.88 | 36.78 |
| trichromy | 40.28 | 14.12 | 107.92 | 37.84 |
These findings imply that the treated water used in a number of dyeing procedures may be recycled, but the preliminary experiments demonstrate that a novel dying method saves 70 % of the dyeing water and significantly lowers the salt of the effluent while reducing consumption expenses. Nevertheless, the energy requirements are partially higher and consequently, the costs could be higher. Therefore, the use of renewable energies is a feasible option to couple with the electrochemical technologies [55,56].
Conclusions
Electrochemical treatment of single and trichromy dye effluents using a parallel plate flow reactor equipped with Ti/Pt or Ti/Pt-Sn-Sb electrocatalytic material has been investigated. Significant decolourization efficiencies were achieved with both electrodes for single dye solutions containing NB CL-R, RY 3RS or RR RR Gran and the trichromy effluent. Higher colour removal was obtained with Ti/Pt-Sn-Sb when compared to the Ti/Pt anode at both current densities, irrespective of the dye effluent treated. The efficiency of EO in decolorizing the dye effluents was influenced by the chemical dye structure in the effluent, as shown by the slow decolourization rate and efficiency of RY 3RS treatment with both electrodes and current densities. COD elimination was hindered during EO treatment of RY 3RS and RR RR Gran due to the formation of polymer films on the anode surface; however, modest COD removal efficiencies of up to 30 % and 74 % were achieved with Ti/Pt and Ti/Pt-Sn-Sb anodes, respectively, for the treatment of trichromy effluents.
The EO process enhanced the DO of the treated effluents and reduced turbidity in most cases owing to the decrease of organic matter and degradation of dye molecules in the effluents. The cell potential was relatively stable during EO of all the dye effluents and a lower potential was observed with Ti/Pt-Sn-Sb compared to the Ti/Pt anode. Lower energy consumption, and in turn, cost, was achieved for the treatment of all dye solutions with Ti/Pt-Sn-Sb compared to the Ti/Pt anode at both current densities. Finally, based on the results reported, EO is an efficient treatment technique for decolorizing industrial effluents containing single and trichromy dyes as well as reducing their ecotoxicological effects and the risk to aquatic environment. However, due to partial or total inhibition of COD removal observed with the electrodes under study, further research is ongoing to investigate the performance of non-active high oxidation potential anodes such as PbO2 and boron-doped diamond for the treatment of trichromy dyes.

